はじめに
新薬開発において「標準治療と比べて十分な有効性を保持しているか」を検証する非劣性試験は、抗菌薬や抗凝固薬、がん領域などで広く用いられています。特に標準治療が確立している領域では、プラセボ対照試験は倫理的に困難であり、非劣性試験が唯一の選択肢となる場合も少なくありません。
しかし、非劣性試験は「どの程度の差まで許容できるか」という非劣性マージンの設定が難しく、統計的・臨床的な妥当性を両立させる必要があります。本記事では、非劣性マージンの設定方法、試験デザインの留意点、さらに規制当局の審査事例を交えて解説します。
非劣性試験と非劣性マージンの基礎
目的:新薬が標準治療に比べて臨床的に意味のある差で劣っていないことを示す。
設定根拠:過去のRCTやメタ解析から標準治療の効果を推定し、保持率(例:50%)を掛け合わせて算出。
非劣性マージン(Δ):標準治療の効果のうち、新薬に最低限保持してほしい割合を定量化したもの。
保持率(Preservation Fraction)の考え方
基本的な定義
- 保持率とは:標準治療がプラセボに対して示した効果のうち、新薬に最低限保持してほしい割合。
- 数式で表すと:
\[\Delta = (\text{効果}_{\text{標準治療}} – \text{効果}_{\text{プラセボ}}) \times (1 – \text{保持率})\]
例:標準治療が10%の絶対リスク低下を示した場合、保持率50%なら新薬は5%以上の効果を保持すべき → Δ = 5%
保持率の設定根拠
保持率は単なる統計的な計算ではなく、臨床的・規制的な妥当性を踏まえて決定されます。
- 臨床的観点
- 効果が多少減弱しても、副作用軽減・利便性(経口投与、投与回数減少など)が大きければ保持率を低めに設定できる。
- 逆に、命に直結する疾患(例:抗がん剤、抗凝固薬)では保持率を高めに設定する必要がある。
- 規制当局の推奨
- FDA:保持率50%を一つの目安とするが、疾患や治療領域に応じて柔軟に判断。
- PMDA:保持率は「臨床的に意味のある効果を確実に保持する」ことを重視し、50%以上を推奨するケースが多い。
実務での保持率設定プロセス
- 標準治療の効果量を推定
- 過去のプラセボ対照試験やメタ解析から効果量を算出。
- 例:イベント発生率が20% → 10%に低下 → 絶対リスク差10%。
- 保持率を決定
- 臨床的意義、副作用プロファイル、利便性を考慮。
- 例:抗菌薬 → 50%保持(Δ = 5%)、抗がん剤 → 67%以上保持(Δ ≈ 3.3%)。
- 規制当局との事前相談
- Δの設定は審査で厳しくチェックされるため、治験開始前にPMDA/FDAと協議することが必須。
具体例
- 抗菌薬
- 標準治療の治癒率:90%、プラセボ:70% → 効果差20%
- 保持率50% → Δ = 10%
- 新薬は治癒率80%以上を示せば非劣性成立
- 抗凝固薬
- 新薬は少なくとも1.34%のリスク低下を保持する必要あり
- ワルファリン vs プラセボ:脳卒中発症率を年間2%低下
- 保持率67% → Δ = 0.66%
非劣性マージン設定の実務プロセス
- 標準治療の効果を定量化
例:プラセボ群20% → 標準治療群10% → 絶対リスク差10% - 保持率を設定
保持率50% → Δ = 5% - 規制当局のガイダンスを参照
- PMDA「非劣性マージンの設定に関する考え方」
- FDA「Non-Inferiority Clinical Trials Guidance」
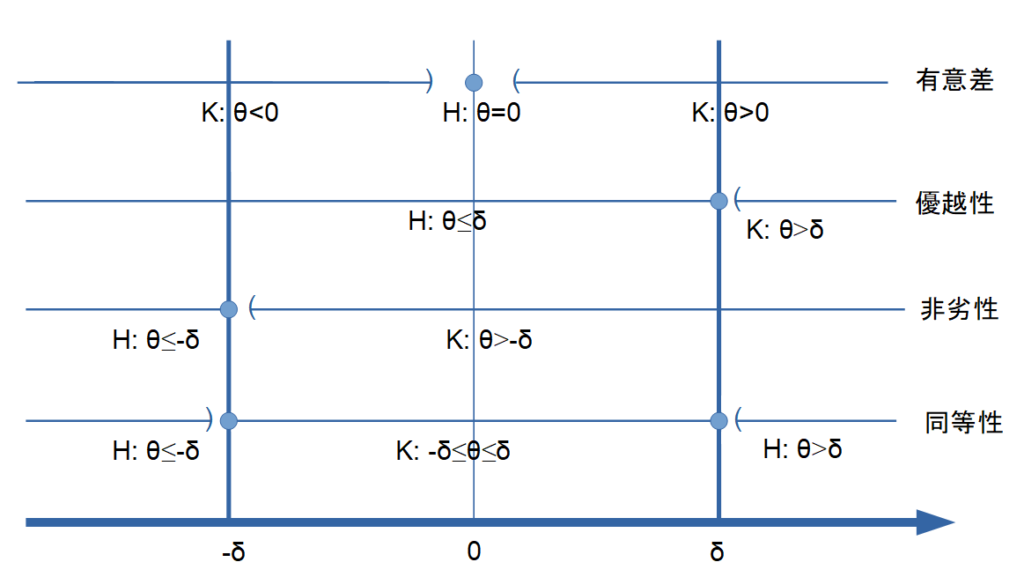
図解で理解する非劣性試験
図解で理解するために、有意差、優越性、非劣性、同等性それぞれの仮説について図式で表すと以下のようになります。
非劣性は臨床的に意味のある\(\sigma>0\)以内であれば臨床的に劣らないとかんがえてよいことを意味しております。
規制当局の審査事例
抗菌薬領域(PMDA事例)
PMDAは抗菌薬の非劣性試験において、「プラセボ対照試験から得られた標準治療の効果を保持すること」を重視しています。ある抗菌薬の承認審査では、Δを過去のメタ解析から算出し、臨床的に意味のある効果を50%以上保持することを条件に非劣性が認められました。
抗凝固薬領域(FDA事例)
FDAは新規経口抗凝固薬(NOAC)の審査において、ハザード比1.38を非劣性マージンとして設定。結果として新薬はHR 0.79(95%CI 0.66–0.95)を示し、非劣性だけでなく優越性も認められました。
この事例は「非劣性試験から優越性が導かれる」典型例として知られています。
実務上の留意点
臨床的意義:統計的非劣性が示されても、副作用や利便性を含めた総合評価が必要。
ITT解析とPP解析:両方で一貫した結果が必要。
サンプルサイズ:Δが小さいほど必要症例数は増加。
規制的観点:PMDAは「非劣性から優越性へのスイッチング」を原則認めていない。
特に4つ目の、非劣性が達成された後に優越性を検証するデザイン(いわゆる,スイッチング)では、多重性の調整はFWERの適切な制御の枠組みに置いては理論上不要であるものの、規制当局間で結果の受け入れ可能性が異なることがあります。
そのため、国際共同試験では、承認申請後に結果の受け入れ可能性について当局間で差異が生じ、議論となることがあるため,試験開始前にこの点についても想定し,当局と十分に議論しておくことが大事となります。
まとめ
非劣性試験は、標準治療が確立しておりプラセボ対照試験が倫理的に実施困難な領域において、極めて重要な試験デザインです。新薬が既存の標準治療に対して臨床的に意味のある差で劣っていないことを示すために用いられます。
この試験において中心的な役割を果たすのが非劣性マージンの設定です。非劣性マージンは、標準治療がプラセボに対して示した効果のうち、新薬に最低限保持してほしい割合(保持率)を掛け合わせて算出されます。すなわち、「標準治療の効果 × 保持率」によって非劣性マージンが導かれます。
実際の審査においては、PMDAやFDAといった規制当局も、非劣性マージンの設定において統計的根拠と臨床的妥当性の両立を強く求めています。過去の審査事例からも、保持率の設定や標準治療の効果推定に対する透明性と合理性が、承認の可否を左右する重要な要素であることが分かります。
さらに、非劣性マージンの大きさは必要なサンプルサイズに直結するため、実施可能性を踏まえた設計が不可欠です。加えて、統計的に非劣性が示されたとしても、それが臨床的に意味のある差であるかどうかを慎重に評価することが、実務上の重要な判断ポイントとなります。