アダプティブデザイン臨床試験の理解と展望
はじめに
新薬開発における臨床試験は、患者の安全性と有効性を科学的に検証するための最も重要なプロセスです。しかし、従来の試験デザインは「固定的」であり、試験開始後に設計を変更することは原則として認められていませんでした。その結果、期待された効果が得られない治療に多くの患者が参加し続ける、あるいはリソースが非効率に消費されるといった課題が生じていました。
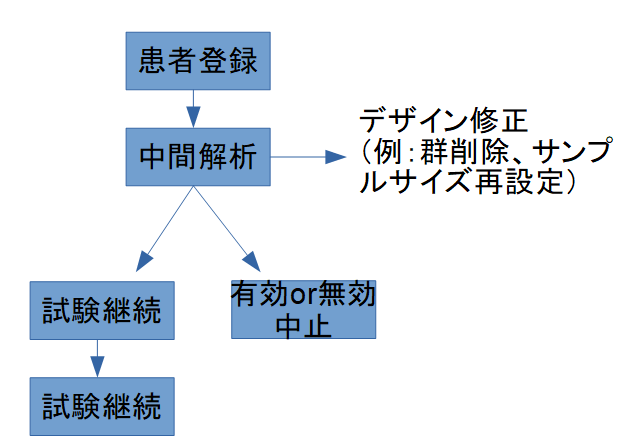
こうした背景のもと登場したのがアダプティブデザイン(Adaptive Design)です。これは、事前に定めたルールに従い、中間解析などで得られた情報を用いて試験の設計を柔軟に変更できる方法論です。アダプティブデザインは、効率性と倫理性を両立させる革新的なアプローチとして注目され、近年ではFDAやEMA、PMDAといった規制当局もその活用に関するガイダンスを整備しつつあります。
本稿では、アダプティブデザインの基本概念、各国規制当局の考え方、統計的課題、実務上の留意点を整理するとともに、Rコードによるサンプルサイズ再設定のシミュレーション例を示します。さらにFDAガイダンスの引用を交え、臨床試験におけるアダプティブデザインの理解を深めることを目的とします。
アダプティブデザインとは
臨床試験は新薬開発の根幹を支えるプロセスですが、従来の試験デザインは「固定的」であり、試験開始後に設計を変更することは原則認められていませんでした。これに対し、アダプティブデザイン(Adaptive Design)は、事前に定めたルールに従い、中間解析などで得られた情報を用いて試験の設計を柔軟に変更できる方法論です。
- 事前計画性:変更のルールは試験開始前に明示される
- 柔軟性:サンプルサイズ、投与群、エンドポイントなどを調整可能
- 効率性:不要な群を早期に削除し、リソースを集中できる
- 倫理性:有効性の低い治療への患者曝露を減らす
規制当局の考え方
FDA(米国)
FDAは2019年に「Adaptive Design Clinical Trials for Drugs and Biologics Guidance」を公表しました。このガイダンスでは以下の点が強調されています:
- 透明性:FDAガイドラインでも下記のような記載の通り、「適応的デザインの臨床試験は、試験の完全性と結果の解釈可能性を確保するために、十分な詳細をもって計画されなければならない」とあります。
「Adaptive design clinical trials should be planned with sufficient detail to ensure the integrity of the trial and the interpretability of the results.」 - 型Ⅰ誤差の制御:試験途中での変更が誤った結論を導かないよう、統計的補正が必須。
- ベイズ統計の活用:事前分布やシミュレーションを用いた柔軟な意思決定を認める。
EMA(欧州)
- 2007年に「Reflection Paper」を公表。
- 確証的試験での利用に慎重姿勢を示しつつ、信頼性確保を強調。
- 型Ⅰ誤差の制御や試験の解釈可能性を最重要視。
PMDA(日本)
- ICH-E20(臨床試験のためのアダプティブデザイン)に参画。
- 国内でも承認事例があり、国際調和のもとガイドライン整備が進展中。
- 規制当局との事前相談が必須。
アダプティブデザインの課題
アダプティブデザインを行う上で以下の課題が考えられます。
第Ⅰ種誤差の制御
試験途中で設計を変更すると、偶然による誤った結論(false positive)のリスクが高まる。これを防ぐために、統計的補正(α調整)が必要。
バイアスのリスク
非盲検データを用いた場合、解析者やスポンサーの判断に影響を与える可能性がある。独立データモニタリング委員会(IDMC)の設置が推奨される。
シミュレーションの重要性
事前に多様なシナリオを想定し、試験の頑健性を確認する必要がある。特に複雑なアダプティブデザインでは、シミュレーションが不可欠。
- 試験運営の複雑化:中間解析のタイミングやデータ管理体制の整備が不可欠。
- 規制当局との対話:事前に適応ルールを明確化し、相談を重ねることが重要。
- 倫理的配慮:患者に不利益を与えないよう、透明性の高い説明責任が求められる。
- リソース負担:統計解析チーム、データマネジメント、モニタリング体制の強化が必要。
Rコードによるサンプルサイズ再設定のシミュレーション例
以下は、効果量が予想より小さい場合にサンプルサイズを再設定するシミュレーション例です。
# サンプルサイズ再設定シミュレーション
set.seed(123)
simulate_trial <- function(n_initial, effect_true, interim_fraction=0.5) {
# 初期サンプルサイズ
n_interim <- round(n_initial * interim_fraction)
# データ生成(正規分布、効果量あり)
data <- rnorm(n_initial, mean=effect_true, sd=1)
# 中間解析
interim_data <- data[1:n_interim]
interim_mean <- mean(interim_data)
# 効果量が小さい場合はサンプルサイズを増加
if (interim_mean < 0.3) {
n_new <- n_initial + 50
} else {
n_new <- n_initial
}
# 最終解析
final_data <- rnorm(n_new, mean=effect_true, sd=1)
p_value <- t.test(final_data)$p.value
return(list(n_final=n_new, p_value=p_value))
}
# シミュレーション実行
results <- replicate(1000, simulate_trial(100, effect_true=0.4), simplify=FALSE)
final_sizes <- sapply(results, function(x) x$n_final)
p_values <- sapply(results, function(x) x$p_value)
# 結果の要約
mean(final_sizes)
mean(p_values < 0.05)
- 初期サンプルサイズを100例と設定。
- 中間解析で効果量が小さい場合、50例追加。
- 最終的な有意差検出率(検出力)を評価。
このようなシミュレーションにより、サンプルサイズ再設定が検出力に与える影響を事前に確認できます。
ケーススタディ
- サンプルサイズ再設定:中間解析で効果量が予想より小さい場合、必要症例数を増加。
- 群削除:有効性が低い投与群を早期に削除し、残りの群に集中。
- 無益性解析:効果が期待できない場合、試験を早期終了。
今後の展望
国際的調和:ICH-E20の最終化により、FDA・EMA・PMDAの共通基盤が整備される。
ベイズ統計の活用:事前分布を用いた柔軟な意思決定が広がる。
実務的成熟:製薬企業やCROにおける運用経験が蓄積され、標準的手法として定着する可能性。
まとめ
アダプティブデザインは、臨床試験の効率化と倫理性向上を両立できる革新的な方法論です。しかし、統計的厳密性・規制当局の承認・実務上の複雑性といった課題を克服する必要があります。FDAガイダンスにあるように「試験の透明性と解釈可能性を確保すること」が最重要であり、シミュレーションや事前計画を通じてその信頼性を担保することが求められます。
今後はICH-E20を中心に国際的なガイドラインが整備され、製薬業界における標準的な選択肢となることが期待されます。