【完全解説】ICH E8(R1)「臨床試験の一般指針」をわかりやすくまとめてみた

はじめに
臨床試験の国際的な標準を定めるICHガイドラインの中でも、E8(臨床試験の一般指針)は、すべての臨床試験の“土台”となる重要文書です。
2021年に改訂された ICH E8(R1) は、従来よりも大きく踏み込み、「臨床試験の質とは何か」を再定義し、クオリティ・バイ・デザイン(Quality by Design; QbD)という新しい考え方を中心に据えています。
この記事では、E8(R1)のポイントを 図解つきでわかりやすく解説します。
臨床開発に関わる方はもちろん、医療者・研究者・学生にも役立つ内容です。
ICH E8(R1)とは?
目的の全体像
E8(R1)は、臨床試験の「質」を以下のように定義します。
“目的への適合性(fitness for purpose)”
= 試験の目的を達成し、信頼できる意思決定を支える情報を得られるかどうか
つまり、「完璧なデータ」よりも「目的に合ったデータ」 が重要だという考え方です。
E8(R1)の目的は次の4つです。
- 参加者保護と規制受容性を両立した臨床試験の原則を示す
- 臨床試験の質を設計段階から作り込むための指針を提供する
- 臨床試験の種類と、それぞれで重要となる質の要因を整理する
- ICHの有効性ガイドライン群へのアクセスを助ける
臨床試験の一般原則
参加者の保護
臨床試験の倫理は ヘルシンキ宣言 を基盤とし、ICH E6(GCP)に沿って実施されるべきとされています。
ヘルシンキ宣言とは、世界医師会が定めた人間を対象とする医学研究の倫理原則で、被験者の人権保護を最重要視し、インフォームド・コンセント(十分な説明と同意)や倫理審査委員会の設置、研究計画の公開などを義務付け、ヒトを対象とする研究の公正性と倫理性を確保するための国際的な指針です
- 治験責任医師・依頼者・IRB/IEC が共同で責任を負う
- 個人情報の機密性を確保
- 試験開始前に安全性情報を十分に収集
- 新たな知見が得られたら試験計画を適宜調整
- 不必要な負担を参加者に課さない
科学的アプローチ
臨床試験は、明確なリサーチクエスチョンに基づき、科学的に妥当な方法でデザイン・実施・解析される必要があります。
- 目的が明確であること
- デザインが目的に適合していること
- データが意思決定に十分な質を持つこと
- 国際共同治験では地域差も考慮(ICH E5/E17)
患者の視点の活用
E8(R1)の大きな特徴が 患者参画(Patient Engagement) の強調です。
患者参画がもたらすメリット
- 組入れ基準の妥当性向上
- 来院スケジュールの負担軽減
- 意義のあるエンドポイント設定
- 試験への信頼性向上 → 組入れ促進・脱落率低下
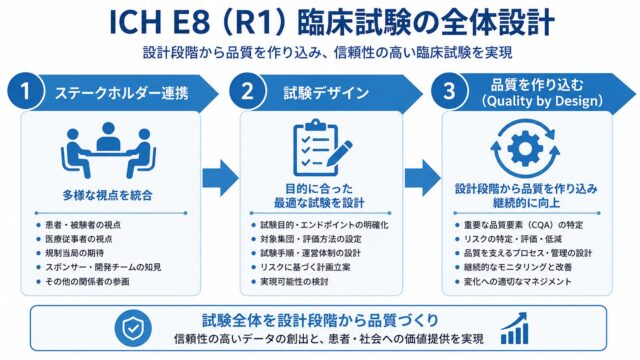
クオリティ・バイ・デザイン(QbD)とは?
E8(R1)の中心概念が QbD(Quality by Design) です。
QbDの考え方
質は“後からチェックするもの”ではなく、“最初から設計するもの”
従来の臨床試験は、
「モニタリング・監査でエラーを見つけて修正する」
という“事後的な質保証”が中心でした。
しかしE8(R1)では、試験計画書・手順・運用設計の段階で質を作り込むことが求められます。
CTQ要因(Critical to Quality Factors)
QbDの核となるのが CTQ要因 です。
試験の目的達成・参加者保護・データ信頼性に重大な影響を与える要因
CTQ要因が損なわれると、試験結果の信頼性や倫理性が揺らぎます。
- 主要評価項目の測定精度・妥当性
- 適切な対象集団の選定
- ランダム化・盲検化の維持
- 重要な安全性イベントの把握
- 来院スケジュールの遵守
- データの完全性(欠測の最小化)
CTQ要因の特定プロセス
E8(R1)は、CTQ要因を特定するためのアプローチを5つ示しています。
開かれた対話の文化
- チェックリスト依存ではなく、批判的思考を重視
- データの矛盾を認め、透明性を確保
- 標準化と柔軟性のバランスを取る
本質的な活動への集中
“やらなくてよいこと”を減らすことが質を高める
- 重要な誤りを防ぐことに注力
- 不要なデータ収集を削減
- リソースを重要な領域に集中
利害関係者の参画
- 患者、医療者、治験コーディネーター、現場スタッフ
- 外部専門家の意見も積極的に取り入れる
- 規制当局との早期相談も推奨
CTQ要因の継続的レビュー
- リスク管理を継続的に更新
- 試験中に新たな問題が発生する可能性
- 中間解析や計画変更時は特に注意
運用上のCTQ要因
実施可能性(Feasibility)はCTQの重要要素。
運用上のCTQ例
- 適格な治験責任医師・CRCの確保
- 必要な設備・検査体制
- 対象患者の確保
- 脱落防止策
- フォローアップ体制
医薬品開発計画(概要)
E8(R1)は、医薬品開発のライフサイクル全体を扱います。
非臨床 → 臨床薬理 → 探索的試験 → 検証的試験 → 承認後試験
各段階で必要な情報が異なり、CTQ要因も変化します。
臨床試験デザインの構成要素
E8(R1)は、臨床試験デザインの重要要素を整理しています。
- 対象集団(患者背景、適格基準)
- 試験治療(用量、投与方法、比較対照)
- 対照群の選択(プラセボ、実薬、外部対照など)
- 反応変数(評価項目)
- バイアス低減手法(ランダム化、盲検化)
- 統計解析(仮説、解析計画、欠測データ対応)
- データソース(ePRO、RWD、レジストリ等)
試験実施・安全性モニタリング・報告
E8(R1)は、試験実施の基本も整理しています。
● 試験実施
- 計画書遵守
- トレーニング
- データマネジメント
- 中間データへのアクセス管理
● 安全性モニタリング
- DMC(データモニタリング委員会)の活用
- 安全性情報の継続的評価
- 中止基準の設定
まとめ
ICH E8(R1)は、臨床試験の質を「目的への適合性」と再定義し、従来の“事後的なチェック中心の品質管理”から、設計段階で質を作り込むクオリティ・バイ・デザイン(QbD)への転換を強く打ち出したガイドラインです。臨床試験の目的を明確にし、その目的を達成するために本当に重要な要素(CTQ要因)を特定し、リスクに応じて管理することが求められます。
CTQ要因は、参加者の保護、データの信頼性、試験結果の解釈可能性に直接影響する要素であり、試験の成功に不可欠です。これらを適切に見極めるためには、患者・医療者・現場スタッフ・規制当局など、多様な利害関係者との対話が重要であり、試験の複雑化を避け、必要な活動に集中する姿勢が求められます。
また、E8(R1)は臨床開発のライフサイクル全体を対象とし、非臨床から承認後試験まで、各段階で必要な情報とデザインの考え方を整理しています。臨床試験デザインの構成要素(対象集団、評価項目、対照群、バイアス低減、統計解析、データソースなど)も体系的に示され、試験の科学的妥当性と実施可能性の両立が強調されています。
総じて、ICH E8(R1)は「より効率的で、より患者中心で、より信頼性の高い臨床試験」を実現するための基盤となるガイドラインです。臨床試験に関わるすべての人が、試験の目的に立ち返り、質の本質を見極めながら設計・実施していくことが求められています。
関連記事
ICHガイドラインを横断的に押さえたい方は、以下の記事もあわせてご覧ください。