ICH-E1ガイドライン解説 ー致命的でない疾患に対する長期投与薬の安全性評価ー

はじめに
新医薬品の開発において、安全性の評価は有効性と並んで最も重要な課題の一つです。特に慢性疾患や生活習慣病など、致命的ではないが長期間の投与が想定される薬剤では、短期的な副作用だけでなく、半年から1年以上にわたる投与によって生じる可能性のある遅発性の有害事象を把握することが不可欠です。
しかし、治験段階で全ての副作用を網羅的に検出することは現実的に困難です。稀な副作用は市販後調査で初めて明らかになることも多く、治験設計においては「どの程度の症例数と投与期間を確保すれば、承認に必要な安全性データベースを構築できるのか」という問いが常に存在します。
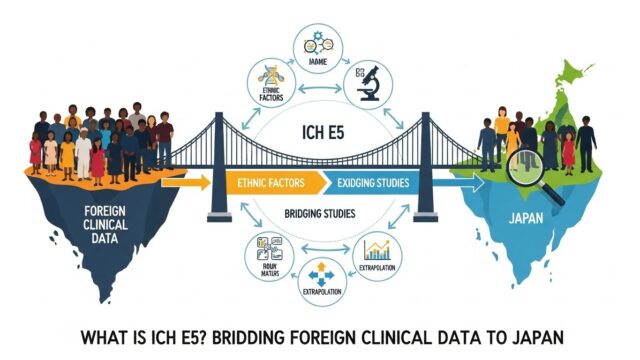
この課題に対応するため、日米EU三極の規制当局が協力して策定したのが ICH-E1ガイドライン です。本ガイドラインは、致命的でない疾患に対して長期投与が想定される新薬の治験段階における安全性評価について、国際的に調和された基準を提示しています。
本記事では、このICH-E1ガイドラインの内容を分かりやすく解説し、製薬企業や研究者が治験計画を立案する際に理解しておくべきポイントを整理します。図解を交えながら、症例数や投与期間の目安、例外的なケース、そして市販後調査との関係について具体的に説明していきます。
ガイドラインの背景
新医薬品の開発においては、承認後に患者へ長期間投与されることが想定される薬剤が少なくありません。特に慢性疾患や生活習慣病の治療薬では、半年から1年以上の継続投与が一般的です。
しかし、治験段階で得られる安全性データは多くの場合短期投与試験に基づいており、長期投与による副作用や遅発性の有害事象を十分に把握できないという課題があります。
この問題に対応するため、ICH(国際医薬品規制調和会議) は「E1ガイドライン」を策定しました。本ガイドラインは、致命的でない疾患に対して長期投与が想定される新薬について、治験段階でどの程度の症例数と投与期間が必要かを示すものです。
基本的な考え方
ガイドラインの中心的な考え方は以下の通りです。
- 有害事象の発現率と投与期間の関係を把握することが重要
→ 多くの副作用は投与開始後数か月以内に発現するが、遅発性の事象も存在するため、6か月以上の観察が必要。 - 稀な有害事象(例:1000例に1件未満)は治験段階での検出は期待されない
→ 市販後調査で補完する。 - 安全性評価のためには、統計学的検出力と実施可能性のバランスが必要
→ 膨大な症例数を求めるのではなく、現実的に収集可能な範囲で設計する。
症例数と投与期間の目安
ガイドラインでは、以下のような目安が示されています。
6か月投与試験
- 症例数:300~600例が適当
- 目的:
- 有害事象の経時的パターンを把握
- 遅発性(0.5~5%程度)の副作用を検出
- 高頻度の副作用が増加するか減少するかを確認

12か月投与試験
- 症例数:100例以上
- 目的:
- 投与開始後6か月以上経って初めて発現する重篤な副作用を確認
- 長期投与による副作用の累積発現率を評価
例:1年間投与で重篤な副作用が認められなければ、累積発現率は3%未満と推定可能。
総症例数
- 500~1500例程度が望ましい
- 米国・EUでは1500例程度が必要とされる。
- 市販後調査でさらに安全性データを収集することが前提。
例外的なケース
ガイドラインは「一般原則」を示していますが、例外も認めています。以下のような場合には、より大規模・長期の安全性データが必要となります。
- 遅発性または累積的に重症化する副作用が予想される薬剤
- 動物試験や類似薬の情報から予測される場合。
- 特定の低頻度の重篤な副作用を推定する必要がある場合
- 類似薬で既に報告されている副作用がある場合。
- 有効性が限定的な薬剤
- 症状改善が軽度、対象患者が少数、代用エンドポイントで評価される場合。
- 背景の罹患率・死亡率が高い場合
- 投与による増加を検出するために十分な統計学的検出力が必要。
- 対象患者数が少ない疾患
- 症例数が少なくても承認可能な場合がある。
実務的なポイント
- 承認申請時点では6か月投与試験の成績で十分
- 12か月投与試験の成績は可能な限り早期に追加提出
- 市販後調査で稀な副作用を補完
この流れにより、承認審査の迅速化と安全性確保の両立を図っています。
ICH-E1ガイドラインは、日米EU三極で調和された基準を提供することで、以下のメリットをもたらします。
- 国際共同治験の効率化
- 承認審査の迅速化
- 患者への早期アクセス
- 安全性評価の透明性向上
まとめ
ICH-E1ガイドラインは、致命的でない疾患に対して長期間の投与が想定される新薬の治験段階における安全性評価について、国際的に調和された基準を示したものです。その目的は、承認後に想定される長期投与に備え、治験段階で必要な症例数と投与期間を明確にすることにあります。
本ガイドラインの基本的な考え方は、有害事象の発現率と投与期間の関係を把握することにあり、短期投与では検出できない遅発性の副作用を確認するために、6か月以上の投与試験が必要とされています。具体的には、
- 6か月投与試験:300~600例
- 12か月投与試験:100例以上
- 総症例数:500~1500例程度
という目安が示されています。これにより、一般的な副作用の発現パターンを把握し、長期投与による安全性を確認することが可能となります。
一方で、遅発性や累積的に重症化する副作用が予想される薬剤、特定の低頻度の重篤な副作用を推定する必要がある薬剤、有効性が限定的な薬剤などについては、より大規模・長期の安全性データベースが必要となる場合もあります。
また、治験段階で検出できない稀な副作用については、市販後調査によって補完することが前提とされています。承認申請時には6か月投与試験の成績で十分とされますが、12か月投与試験の成績は可能な限り早期に追加提出することが求められています。
総じて、ICH-E1ガイドラインは、安全性評価のための症例数と投与期間を国際的に統一することで、治験設計の効率化、承認審査の迅速化、患者への早期アクセスを実現することを目的としています。製薬企業や研究者にとっては、治験計画を立案する際の重要な指針であり、規制当局にとっても安全性評価の透明性を高める基盤となるものです。
ICHガイドラインについてさらに詳細に勉強したい方は下記資料集がおすすめです。

![[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]](https://hbb.afl.rakuten.co.jp/hgb/4aa03195.892069bb.4aa03196.39df6be9/?me_id=1213310&item_id=19096259&pc=https%3A%2F%2Fthumbnail.image.rakuten.co.jp%2F%400_mall%2Fbook%2Fcabinet%2F0677%2F9784840750677.jpg%3F_ex%3D400x400&s=400x400&t=picttext "[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]")