CTD(コモン・テクニカル・ドキュメント)とは ― 5モジュール構成と医薬品承認申請の全体像を生物統計家の視点で徹底解説 ―

・CTD(コモン・テクニカル・ドキュメント)とは何か、ICH M4 が定める国際共通様式の意味
・CTD を構成する 5 つのモジュール(Module 1〜5)それぞれの役割と中身
・eCTD による電子申請の流れと、紙申請からの転換
・CTD のどの部分に生物統計家が関わるのか(CTD 2.5/2.7・Module 5)
・日本(PMDA)への承認申請ならではの注意点
はじめに
新しい医薬品を患者さんのもとへ届けるためには、規制当局による承認審査を通過する必要があります。その際に提出する資料は、品質・非臨床・臨床にわたって膨大な量にのぼり、かつては日本・米国・欧州でそれぞれ異なる様式で作成しなければなりませんでした。同じ試験データを国ごとに作り直す負担は、開発の遅れとコストの増大に直結していたのです。
この非効率を解消するために整備されたのが CTD(Common Technical Document:コモン・テクニカル・ドキュメント) です。CTD は医薬品規制調和国際会議(ICH)で合意された「申請資料の国際共通様式」であり、今日では新薬から後発医薬品まで、日本における承認申請の標準フォーマットとなっています。
本記事では、製薬企業で承認申請・臨床開発・生物統計に携わる方や、これから医薬品開発を学ぶ学生の方に向けて、CTD の全体像を体系的に整理します。5 つのモジュールの構成から、eCTD による電子申請、そして生物統計家が関わる臨床のまとめ文書まで、図解を交えてわかりやすく解説します。
CTD とは何か ― ICH M4 が定める申請資料の国際共通様式
CTD とは、医薬品の承認申請に必要な資料を、日米 EU(三極)で共通の構成・順序にまとめた国際共通様式のことです。その構成を規定しているのが ICH の品質・安全性・有効性を横断する「M4(複合領域)ガイドライン」です。M4 は「どのような順番で、どの情報を、どの章立てで提出するか」という資料の“器”を定めるものであり、CTD はその器に沿って作成された申請資料一式を指します。
CTD が登場する以前は、同一の開発データであっても各極の様式に合わせて再編集する必要がありました。CTD によって構成が共通化されたことで、申請者は一度作成した資料を三極で使い回しやすくなり、審査当局も同じ章立てで効率的にレビューできるようになりました。これは国際同時開発を後押しする大きな基盤になっています。
日本では 2001 年に CTD に関する通知が発出され、2003 年 7 月以降の新有効成分含有医薬品の承認申請で CTD が必須となりました。その後、適用範囲はバイオテクノロジー応用医薬品へ、さらに後発医薬品の承認申請へと拡大しています。
CTD は「申請資料の構成・見せ方」を定めた様式であり、実施すべき試験の中身そのものを規定するものではありません。どのような試験を行い、どう評価するかは品質(Q)・安全性(S)・有効性(E)の各 ICH ガイドラインが定めます。CTD はそれらの成果を“共通の器”に整理するための枠組み、と理解すると混乱しません。
なお、医薬品開発の全体像(臨床試験の一般原則)については、ICH の上位指針も押さえておくと理解が深まります。あわせて 【完全解説】ICH E8(R1)「臨床試験の一般指針」 もご参照ください。
CTD の全体像 ― ピラミッド型の 5 モジュール構成
CTD は 5 つのモジュール(Module 1〜5) で構成されます。よく「ピラミッド(三角形)」の図で表現され、頂点に近いほど要約された情報、底辺に近いほど詳細な一次データが配置されるイメージです。
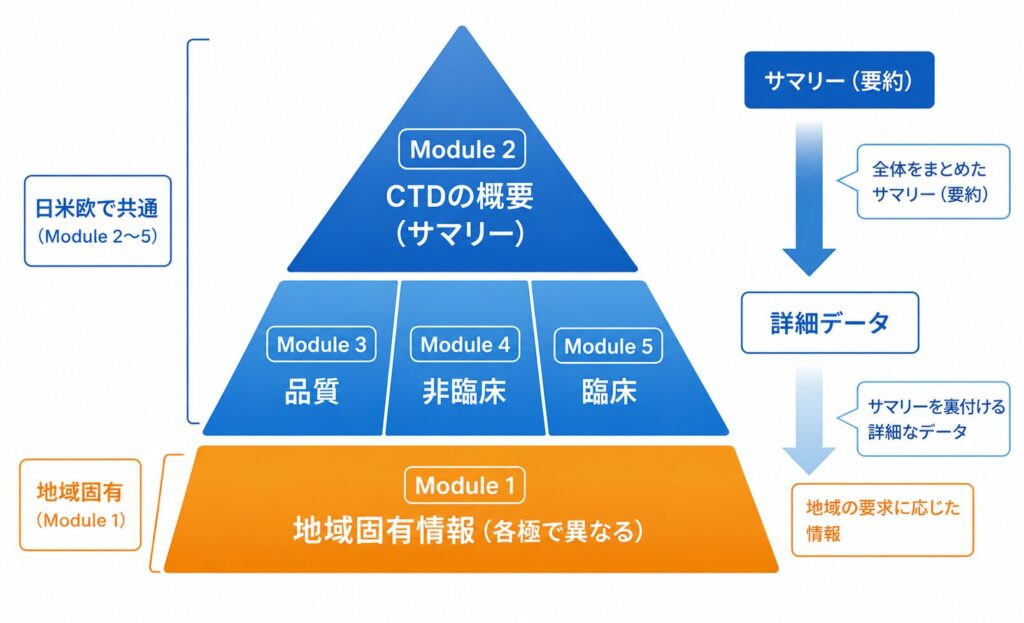
ポイントは、Module 1 だけが各極(日本・米国・欧州)で内容が異なる「地域固有情報」であり、Module 2〜5 は三極で共通になるよう意図されている点です。つまり、Module 2 以降は一度作り込めば国際的に活用でき、各国対応は主に Module 1 で行う、という分業構造になっています。
| モジュール | 名称 | 共通/地域固有 | 主な内容 |
|---|---|---|---|
| Module 1 | 地域固有情報 | 地域固有 | 申請書、添付文書(案)、特許・GMP 関連など各極ごとの行政資料 |
| Module 2 | CTD の概要(サマリー) | 共通 | 品質・非臨床・臨床の概括評価と要約(審査官が最初に読む中核) |
| Module 3 | 品質に関する文書 | 共通 | 原薬・製剤の CMC(製造方法・規格・安定性など) |
| Module 4 | 非臨床試験報告書 | 共通 | 薬理・薬物動態・毒性試験(GLP)の報告書 |
| Module 5 | 臨床試験報告書 | 共通 | 各臨床試験の総括報告書(CSR)、症例一覧、解析データ |
各モジュールの詳細
Module 1:地域固有の情報
Module 1 は、各極の規制要件に応じた行政的な資料を収める領域です。日本であれば承認申請書、添付文書(案)、製造販売業の許可関連資料、特許・再審査期間に関する情報などが含まれます。ここは三極で共通化されておらず、申請先の国・地域ごとに作り分ける必要があります。国際開発では「Module 2〜5 は共通基盤、Module 1 で各国対応」という整理が実務上の基本になります。
Module 2:CTD の概要(サマリー)
Module 2 は、品質・非臨床・臨床の全体を要約したサマリーであり、審査官が最初に通読する CTD の“顔”にあたります。ここでの記述の質が審査の第一印象を左右するため、作成には特に注力されます。Module 2 はさらに 2.1〜2.7 に細分化されます。
| 番号 | 名称 | 内容の要点 |
|---|---|---|
| 2.1 / 2.2 | 目次・緒言 | CTD 全体の目次と、医薬品の概略・効能効果の紹介 |
| 2.3 | 品質に関する概括資料(QOS) | Module 3(品質)の要約 |
| 2.4 | 非臨床に関する概括評価 | 非臨床データの統合的な考察 |
| 2.5 | 臨床に関する概括評価 | ベネフィット・リスクを論じる臨床の最重要サマリー |
| 2.6 | 非臨床試験の概要文・概要表 | 薬理・PK・毒性の文章サマリーと一覧表 |
| 2.7 | 臨床概要 | 有効性(2.7.3)・安全性(2.7.4)など臨床データの詳細な要約 |
特に 2.5(臨床概括評価) と 2.7(臨床概要) は、生物統計家が深く関わるパートです。有効性の統合解析や安全性のまとめは、ここで臨床開発チームと協働して作成されます。臨床試験の統計的な評価軸については 【完全理解】ICH E9「臨床試験の統計的原則」と補遺(Estimand) が基礎になります。
Module 3:品質に関する文書(CTD-Q)
Module 3 は、原薬および製剤の品質(CMC:Chemistry, Manufacturing and Control)に関する詳細データを収めます。製造方法、規格および試験方法、安定性試験の結果などが含まれ、品質の一貫性と再現性を裏づける領域です。バイオ医薬品では、製造工程の管理や同等性/同質性の評価がとりわけ重要になります。
Module 4:非臨床試験報告書
Module 4 には、薬理試験、薬物動態(PK)試験、毒性試験など、ヒト以外を対象とした非臨床試験の報告書が収められます。これらの多くは GLP(医薬品の安全性試験の実施に関する基準)に準拠して実施され、ヒトへの投与に進む前の安全性の根拠を提供します。
Module 5:臨床試験報告書(CSR)
Module 5 は、実施した各臨床試験の 総括報告書(CSR:Clinical Study Report) を中心とする、臨床データの“一次情報”が集約された領域です。CSR の構成は ICH E3ガイドライン(治験総括報告書の構成と内容) に従って標準化されており、試験デザイン、統計解析計画、解析結果、症例一覧などが体系的にまとめられます。
生物統計家にとって Module 5 は、統計解析計画書(SAP)、解析結果、解析データセットの位置づけを理解するうえで中核となるパートです。近年は解析用データセット(ADaM など)の電子提出も一般化しており、データの再現性・追跡可能性が重視されています。データセット作成の実務については ADaMデータセット自動生成とAI活用 もあわせてご覧ください。
日本(PMDA)申請の実務と eCTD
CTD の提出は、現在では電子形式である eCTD(electronic Common Technical Document) が標準です。eCTD は CTD の構成を電子的に表現し、各文書をハイパーリンクやライフサイクル管理(差し替え履歴の管理)とともに提出できる仕組みで、日本では PMDA のゲートウェイシステムを通じて提出します。紙の申請資料に比べ、審査側の参照性・検索性が大きく向上します。
Module 1 は日本固有の行政資料であり、PMDA との対面助言(治験相談)で論点を事前にすり合わせておくことが、申請後の照会事項を減らすうえで有効です。また、提出データは 信頼性の基準(資料の正確性・完全性・保存)を満たす必要があり、解析結果のトレーサビリティが問われます。
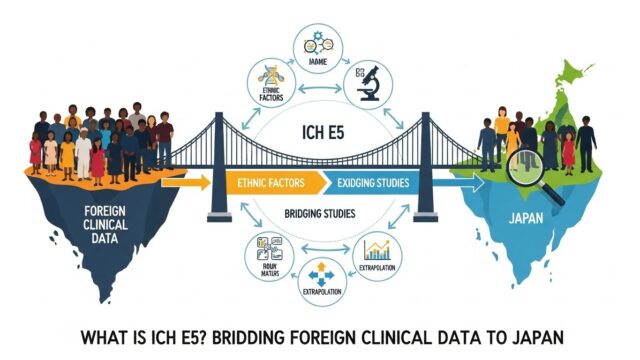
国際共同治験(MRCT)で得たデータを日本の申請に用いる場合は、民族的要因の評価や日本人データの位置づけが論点になります。これらの考え方は 【完全解説】ICH E5(外国臨床データの活用・ブリッジング) や 【徹底解説】ICH E17ガイドライン:国際共同治験(MRCT) で詳しく解説しています。さらに、CTD 全体の信頼性を担保する臨床試験の品質管理の枠組みとして ICH E6(GCP)とは?改訂のポイントまで図解で解説 も重要です。
CTD と生物統計家の関わり
CTD は規制・薬事の文書という印象が強いですが、実際には生物統計家が深く関与するパートが複数あります。とりわけ臨床の有効性・安全性のまとめは、統計解析の結果を“当局が読める形”に翻訳する作業そのものです。
| CTD の場所 | 文書 | 生物統計家の主な関与 |
|---|---|---|
| 2.5 | 臨床概括評価 | ベネフィット・リスク評価の統計的根拠の提示 |
| 2.7.3 | 有効性の概要 | 主要・副次評価項目の統合解析、部分集団解析 |
| 2.7.4 | 安全性の概要 | 有害事象の集計・統合、曝露量との関連の整理 |
| Module 5 | CSR・SAP・ADaM | 統計解析計画の策定、解析の実施、解析データセットの整備 |
近年は、こうした有効性・安全性の評価を「何を推定したいのか」から明確化する Estimand の考え方が重視されています。CTD 2.7 や CSR の解析設計にも直接影響するため、ICH E9(R1) Estimandフレームワーク徹底解説 もあわせて理解しておくと、CTD 作成の質が高まります。
📚 この記事をより深く理解するための参考書籍
CTD・医薬品開発・臨床統計をさらに深く学びたい方に、おすすめの書籍をご紹介します。






関連記事
CTD と関わりの深い ICH ガイドラインや臨床統計のトピックは、以下の記事で詳しく解説しています。あわせて読むことで、承認申請の全体像がより立体的に理解できます。
- 【完全解説】ICH E8(R1)「臨床試験の一般指針」
- 【完全理解】ICH E9「臨床試験の統計的原則」と補遺(Estimand)
- ICH E3ガイドライン徹底解説:治験総括報告書(CSR)の構成と内容
- ICH E6(GCP)とは?改訂のポイントまで図解でわかりやすく解説
まとめ
本記事では、医薬品の承認申請に用いる国際共通様式 CTD(コモン・テクニカル・ドキュメント) について、ICH M4 が定める背景から、5 つのモジュールの構成、eCTD による電子申請、そして生物統計家が関わる臨床のまとめ文書までを体系的に整理しました。
CTD は単なる書類の束ではなく、品質・非臨床・臨床という膨大なエビデンスを「当局が効率的に審査できる共通の器」に整える仕組みです。Module 2〜5 を国際共通の基盤として作り込み、Module 1 で各国対応を行うという構造を理解すれば、国際同時開発の全体像も見通しやすくなります。
とりわけ生物統計家にとっては、CTD 2.5/2.7 や Module 5 が、解析結果をエビデンスとして提示する最終工程です。解析計画の段階から CTD の構成を意識しておくことが、説得力のある申請資料づくりと、審査の円滑化につながります。CTD と関連の深い ICH ガイドラインの記事も参照しながら、承認申請の理解を一段深めていただければと思います。