生物学的同等性試験(BE)とは ― 2×2クロスオーバー設計と90%信頼区間による同等性評価をRで実装する ―

この記事でわかること
・生物学的同等性(BE)とは何か、なぜジェネリック医薬品の承認に「有効性の再証明」ではなく「PK的同等性」が求められるのかがわかります
・主要評価項目となる薬物動態(PK)パラメータ ― AUC(曝露量)と Cmax(最高血中濃度)― の意味と役割が整理できます
・標準的な 2×2 クロスオーバー設計(2製剤・2期間・2順序)の仕組みと、被験者内比較が高い検出力を生む理由が理解できます
・PKデータをなぜ対数変換してから解析するのか、そして同等性マージン 0.80〜1.25 がどこから来るのかを説明できるようになります
・90%信頼区間を用いた同等性評価の考え方と、Rでの実装の全体像をつかめます
はじめに
医療費の抑制が国家的な課題となるなか、特許の切れた先発医薬品と同じ有効成分を含むジェネリック医薬品(後発医薬品)は、患者さんの自己負担と薬剤費全体を下げる重要な役割を担っています。先発品の開発には膨大な臨床試験と費用がかかりますが、後発品はその有効性・安全性の知見を活用できるため、薬価を大きく抑えられます。
ここで一つの疑問が生じます。後発品はあらためて「効くこと」を大規模な臨床試験で示す必要があるのでしょうか。答えは「いいえ」です。有効成分が同じであれば、体内に同じように薬が入り、同じように分布・代謝・排泄される ― すなわち血中濃度の推移が先発品と「同等」であれば、臨床効果も同等とみなせると考えます。この「同等であること」を統計的に示す試験が、本記事のテーマである生物学的同等性試験(Bioequivalence study, BE試験)です。
本記事は、製薬企業やCRO(医薬品開発受託機関)でBE試験に関わる生物統計担当者、そして薬学・統計学を学ぶ学生の方を主な読者として想定しています。前半で生物学的同等性の概念とPK指標、デザインの考え方を丁寧に解説し、後半ではRを用いた具体的な解析手順までを通して学べる構成です。
生物学的同等性とは ― ジェネリック医薬品とPK指標
生物学的同等性(bioequivalence)とは、有効成分が同じ2つの製剤を同じ条件で投与したときに、その薬物動態(pharmacokinetics, PK)が統計的に同等とみなせることを指します。ここで鍵になるのが生物学的利用能(バイオアベイラビリティ, bioavailability)という概念です。これは、投与した薬がどれだけの量・速さで全身循環血(血液)に到達したかを表す指標で、薬の効き目を左右する直接の決定因子です。先発品と後発品でバイオアベイラビリティが揃っていれば、作用部位に届く薬の量とタイミングも揃い、臨床効果も等しくなると考えられます。
BE試験では、先発品を「標準製剤(Reference, R)」、評価したい後発品を「試験製剤(Test, T)」と呼びます。試験の目的は「Tの有効性がRより優れているか」を示すことではなく、あくまで「TがRと同等の範囲に収まっているか」を示すことにあります。優越性試験とは発想がまったく異なる点に注意してください。
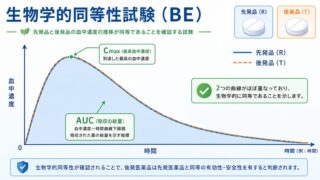
バイオアベイラビリティは、血中濃度を時間の経過に沿って測定した「血中濃度–時間曲線」から評価します。代表的なPKパラメータは次の3つです。
| PKパラメータ | 意味 | BE試験での位置づけ |
|---|---|---|
| AUC (曲線下面積) | 血中濃度–時間曲線の下の面積。体内に取り込まれた薬の総「曝露量」を表す | 主要評価項目(曝露の大きさ) |
| Cmax (最高血中濃度) | 観測された血中濃度のピーク値。薬が「どれだけ速く・高く」入るかを表す | 主要評価項目(曝露の速さ) |
| Tmax (最高血中濃度到達時間) | Cmax に到達するまでの時間。吸収の速さの目安 | 参考項目(統計的同等性評価の対象外とすることが多い) |
AUC(area under the curve, 曲線下面積)は、体に取り込まれた薬の総量、すなわち「曝露量」を表します。AUCが大きいほど、トータルで多くの薬が全身を巡ったことを意味します。一方 Cmax(最高血中濃度)は血中濃度のピークの高さで、薬がどれだけ速く・高く立ち上がるかを示します。AUCが「どれだけの量が入ったか」、Cmaxが「どれだけ速く入ったか」を担うため、この2つを揃えれば曝露の量と速さの両面で先発品を再現できると考えられ、BE試験の主要評価項目とされます。
Tmax(最高血中濃度到達時間)は吸収速度の目安にはなりますが、離散的な測定時点に依存しばらつきが大きいため、統計的な同等性評価の主要項目には通常用いず、参考情報として扱います。
生物学的同等性試験のデザイン ― 2×2クロスオーバーと対数変換
BE試験で最も標準的に用いられるのが、2×2クロスオーバー設計(two-treatment, two-period, two-sequence crossover design)です。「2×2」とは、2つの製剤(T と R)を、2つの期間(第1期・第2期)にわたって、2つの投与順序(TR と RT)に割り付けることを意味します。被験者は無作為に2つの順序群のいずれかに振り分けられ、一方の群は第1期にT・第2期にR、もう一方の群は第1期にR・第2期にTを服用します。
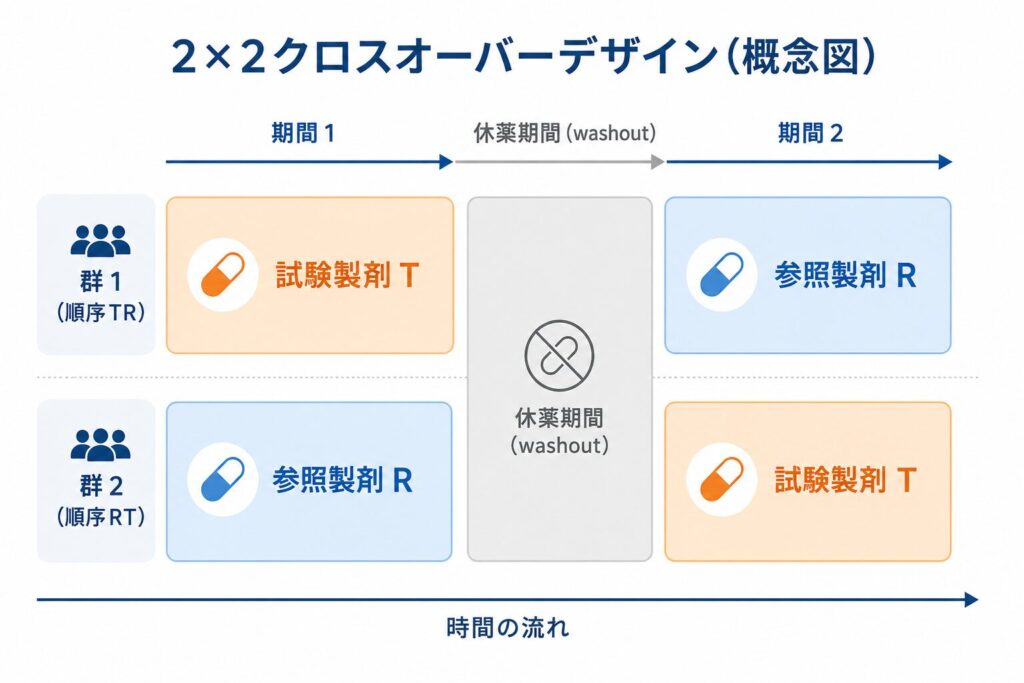
割付の構造を表にすると、次のように整理できます。
| 順序群 | 第1期 | 休薬期間 | 第2期 |
|---|---|---|---|
| 順序1(TR) | 試験製剤 T | washout | 標準製剤 R |
| 順序2(RT) | 標準製剤 R | washout | 試験製剤 T |
このデザインの最大の利点は、同じ被験者がTとRの両方を服用するため、製剤の差を被験者内比較(within-subject comparison)で評価できる点にあります。薬の吸収や代謝には大きな個人差がありますが、同じ人の中でTとRを比べれば、その個人差はキャンセルされます。結果として誤差が小さくなり、被験者間比較(並行群間試験)よりも少ない例数で高い検出力を得られます。BE試験で健康成人を対象とするのも同じ発想で、疾患や併用薬による曝露のばらつきを排し、製剤そのものの差を鋭く検出するためです。
2つの期間の間には十分な休薬期間(washout, ウォッシュアウト)を設ける必要があります。これは、第1期に投与した薬が体内からほぼ消失してから第2期に入るためで、目安として有効成分の半減期の5倍以上が推奨されます。washoutが不足すると、前の期間の薬が次の期間に残るキャリーオーバー効果(持ち越し効果)が生じ、製剤間の比較がゆがんでしまいます。
もう一つの重要なポイントが、解析の前にPKデータを対数変換(log変換)することです。AUCやCmaxといったPKパラメータは値が必ず正で右に裾を引く分布をとり、対数正規分布によく従うことが経験的に知られています。対数をとることで分布が正規分布に近づき、線形モデルの前提(正規性・等分散性)が満たされやすくなります。
さらに、対数変換には比の評価を差の評価に変換できるという数学的な利点があります。BE評価で本当に知りたいのは T と R の平均の「比」 \( \mu_T / \mu_R \) ですが、対数をとると次のように差に置き換えられます。
\[ \ln\!\left(\frac{\mu_T}{\mu_R}\right) = \ln(\mu_T) – \ln(\mu_R) \]
ここで \( \mu_T \) は試験製剤、\( \mu_R \) は標準製剤の母平均を意味します。比の推定は扱いが難しい一方、差の推定は線形モデルや t 分布の枠組みでそのまま扱えるため、解析が格段に容易になります。
最後に同等性マージンについてです。BE試験では、T と R の幾何平均比が原則として 0.80〜1.25 の範囲に収まれば「同等」と判定します。一見すると上下が非対称に見えますが、これは比のスケールで考えると自然です。なぜなら \( 1/1.25 = 0.80 \) であり、対数スケールに移すと中心0に対して対称な区間になるからです。
\[ \ln(0.80) \le \mu_T – \mu_R \le \ln(1.25) \]
ここで \( \ln(1.25) \approx 0.2231 \)、\( \ln(0.80) \approx -0.2231 \) であり、対数スケールでは \( \pm\ln(1.25) \) という0を中心とした対称区間になります。この区間の中に T と R の差の信頼区間がすっぽり収まれば、両製剤は同等と結論づけます。次章以降では、この同等性をどのように90%信頼区間を使って判定し、Rで実装していくのかを具体的に見ていきます。
統計的評価の枠組み ― TOSTと90%信頼区間
生物学的同等性(BE)試験で最初につまずきやすいのが、「同等性を示す」という目標と、私たちが学校で習った検定の論理が真逆だという点です。通常の仮説検定では「差がない(μT − μR = 0)」を帰無仮説に置き、これを棄却して「差がある」と主張します。しかしBEでは、この枠組みをそのまま使うと困ったことが起こります。サンプルサイズが小さく検出力が低いほど帰無仮説が棄却されにくくなり、「差を検出できなかった=同等だ」と結論されてしまうのです。検出力の低い杜撰な試験ほど同等と判定されやすい、という本末転倒な事態になります。
そこでBEでは発想を逆転させます。帰無仮説を「製剤間に意味のある差がある(差が同等域の外にある)」とし、これを棄却できたときに初めて「同等である」と積極的に主張するのです。「差がないことを証明する」のではなく、「差が許容範囲を超えているという仮説を棄却する」という形に組み替えるわけです。
BEでは帰無仮説と対立仮説が通常の検定と逆になります。帰無仮説=「同等でない(差が同等域の外)」、対立仮説=「同等である(差が同等域の内)」。この逆転を理解することが、TOSTと90%信頼区間を腑に落とすための出発点です。
TOST(2つの片側検定)の考え方
同等域は対数スケールで考えます。規制上の同等域は幾何平均比で 0.80〜1.25 と定められており、これを自然対数に変換すると、下限 \( \ln(0.80) = -0.2231 \)、上限 \( \ln(1.25) = +0.2231 \) となります。0.80 と 1.25 が互いに逆数(\(1/0.80 = 1.25\))であるため、対数スケールでは原点 0 をはさんで左右対称の区間 \([-0.2231,\ +0.2231]\) になる点が、対数変換を用いる大きな理由のひとつです。
TOST(Two One-Sided Tests, 2つの片側検定)は、この同等域の「下側にはみ出していないか」と「上側にはみ出していないか」を、それぞれ独立した片側検定として評価する方法です。対数スケールの処置差を \( \delta = \mu_T – \mu_R \) とすると、次の2つの帰無仮説を立てます。
\[ H_{01}:\ \delta \le \ln(0.80) \qquad H_{02}:\ \delta \ge \ln(1.25) \]
ここで \(H_{01}\) は「試験製剤が下限を割り込むほど低い」、\(H_{02}\) は「試験製剤が上限を超えるほど高い」という、それぞれ同等でない側の主張です。この両方をともに有意水準 5%(片側 α = 0.05)で棄却できたときに限り、「差は同等域の内側にある=同等である」と結論します。片側どちらか一方でも棄却できなければ、同等とは言えません。
それぞれの検定統計量は、対数スケールの点推定 \(\hat\delta\) とその標準誤差 \(SE\) を用いて次のように書けます。
\[ t_L = \frac{\hat\delta – \ln(0.80)}{SE}, \qquad t_U = \frac{\hat\delta – \ln(1.25)}{SE} \]
\(t_L\) が自由度 \(\nu\) の t 分布の上側 5% 点を上回り、かつ \(t_U\) が下側 5% 点を下回れば、両方の片側仮説を棄却したことになります。
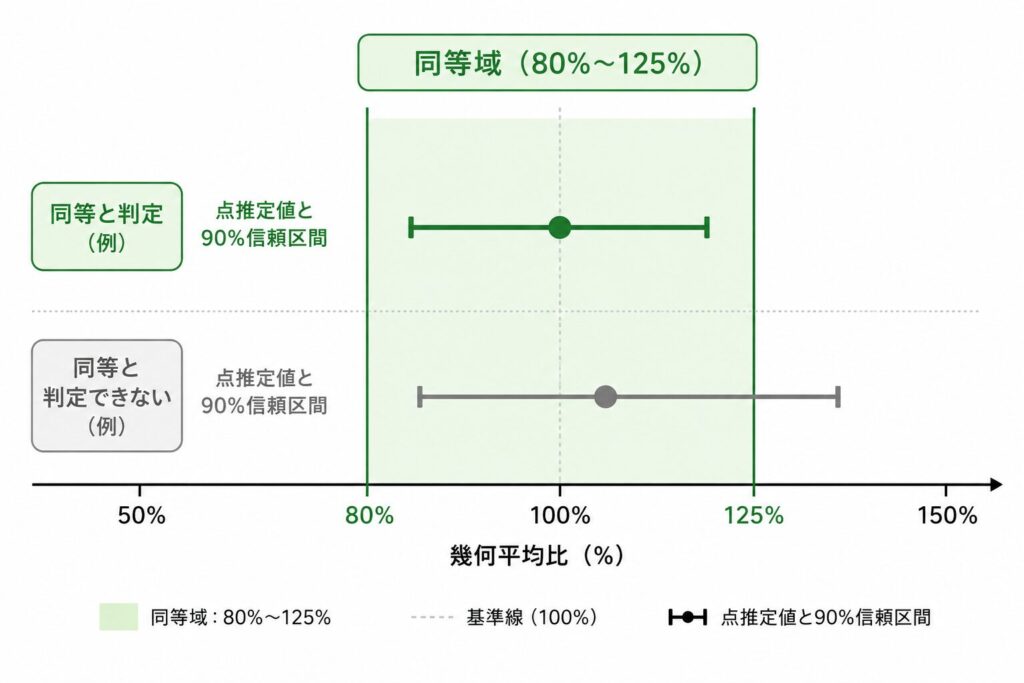
なぜ95%でなく90%信頼区間なのか
TOSTは2つの片側検定の組み合わせですが、実務では同じ結論を信頼区間で表現するのが一般的です。両者は数学的に完全に等価で、幾何平均比(GMR)の90%信頼区間が同等域 [0.80, 1.25] に完全に含まれていれば、TOSTで同等と結論することと一致します。
ここで多くの人が疑問に思うのが、「なぜ95%ではなく90%なのか」という点です。鍵は、TOSTが片側 α = 0.05 の検定を「2つ」行っている点にあります。区間推定の言葉に翻訳すると、両側でそれぞれ 5% ずつを切り落とした区間、すなわち中央の 100% − 2×5% = 90% を信頼区間とすることが、片側 5% の2つの検定とちょうど対応します。両側 95% 信頼区間は片側 2.5% の検定に対応するため、ここでは過剰に厳しすぎる(実際には片側 5% で十分な)ことになります。つまり、規制で定められた検定の厳しさ(片側5%)に整合する区間が90%信頼区間なのです。
BE試験で報告すべきは「90%信頼区間」です。日常的な群間比較の感覚で95%信頼区間を計算してしまうと、TOSTの片側5%とは対応せず、判定基準が変わってしまいます。GMRの点推定が1.00に近くても、90%信頼区間が80〜125%の外にはみ出していれば同等とは結論できません。
解析モデルと幾何平均比
対数変換した PK パラメータに対して、線形モデル(混合モデルまたは分散分析)を当てはめます。標準的な2×2クロスオーバー試験では、固定効果として処置(treatment:T か R)・期間(period:第1期か第2期)・順序(sequence:TR か RT)を、変量効果として順序内の被験者(subject)を置きます。各被験者が両製剤を服用するクロスオーバーデザインのため、製剤間の比較は被験者内で行われ、被験者間のばらつきの影響を取り除いた精度の高い推定が可能になります。標準誤差はこの被験者内変動から求められます。
このモデルから得られる処置効果の推定値 \(\hat\delta = \hat\mu_T – \hat\mu_R\) は対数スケールの差です。これを指数変換すると幾何平均比(GMR)になります。
\[ \text{GMR} = \exp(\hat\delta) = \exp(\hat\mu_T – \hat\mu_R) \]
90%信頼区間も同様に、対数スケールで区間を求めてから指数変換します。
\[ \left[\exp\!\left(\hat\delta – t_{0.95,\nu}\,SE\right),\ \exp\!\left(\hat\delta + t_{0.95,\nu}\,SE\right)\right] \]
ここで \( t_{0.95,\nu} \) は自由度 \(\nu\) の t 分布の上側 5% 点(両側でみれば中央90%を残す値)です。対数スケールでは差の信頼区間として左右対称ですが、指数変換すると比の信頼区間になり、点推定 GMR を中心にやや非対称な区間として得られます。この指数変換後の区間が [0.80, 1.25]、パーセント表示で [80%, 125%] に収まっているかどうかが、同等性判定そのものになります。
RによるBE解析の実装
ここからは、実際のBE解析の流れをRで再現します。データ作成、対数変換した線形混合モデルの当てはめ、処置効果からのGMRと90%信頼区間の算出、そして同等性の判定という順に進めます。
まず、2×2クロスオーバー試験のサンプルデータを作成します。被験者を TR 群(第1期にT、第2期にR)と RT 群(第1期にR、第2期にT)に分け、AUCを対数正規分布から生成します。真のGMRを約0.95、被験者内変動係数(CV)を約0.2 に設定し、被験者間のばらつきも加えて現実的なデータにします。
# 必要なパッケージ
library(nlme)
set.seed(2026)
n_per_seq <- 24 # 各順序群の被験者数
subjects <- 1:(2 * n_per_seq) # 全48名
# 被験者ごとのランダム効果(被験者間変動)
subj_effect <- rnorm(length(subjects), mean = 0, sd = 0.30)
# 真値の設定(対数スケール)
mu_R <- log(100) # 標準製剤Rの対数平均
delta <- log(0.95) # 真の処置差 T - R (GMR=0.95)
cv_w <- 0.20 # 被験者内CV
sd_w <- sqrt(log(cv_w^2 + 1)) # CVから被験者内SDへ変換
# 順序の割り付け
seq_label <- rep(c("TR", "RT"), each = n_per_seq)
make_obs <- function(subj, period, trt) {
mu_trt <- mu_R + ifelse(trt == "T", delta, 0)
log_val <- mu_trt + subj_effect[subj] + rnorm(1, 0, sd_w)
exp(log_val)
}
dat <- do.call(rbind, lapply(seq_along(subjects), function(i) {
s <- subjects[i]
sq <- seq_label[i]
p1_trt <- ifelse(sq == "TR", "T", "R") # 第1期の製剤
p2_trt <- ifelse(sq == "TR", "R", "T") # 第2期の製剤
rbind(
data.frame(subject = s, sequence = sq, period = 1, treatment = p1_trt,
AUC = make_obs(s, 1, p1_trt)),
data.frame(subject = s, sequence = sq, period = 2, treatment = p2_trt,
AUC = make_obs(s, 2, p2_trt))
)
}))
dat$subject <- factor(dat$subject)
dat$sequence <- factor(dat$sequence)
dat$period <- factor(dat$period)
dat$treatment <- factor(dat$treatment, levels = c("R", "T")) # Rを基準
head(dat)
subject sequence period treatment AUC
1 1 TR 1 T 92.41037
2 1 TR 2 R 101.85622
3 2 TR 1 T 88.07719
4 2 TR 2 R 95.32048
5 3 TR 1 T 110.64513
6 3 TR 2 R 118.79204
各被験者(subject)が第1期(period=1)と第2期(period=2)で異なる製剤を服用する、典型的な2×2クロスオーバーの構造になっています。TR群の被験者1は第1期にT、第2期にRを服用していることが確認できます。treatmentの基準をR(標準製剤)に設定したので、後で取り出す処置効果は「T − R」として解釈できます。
次に、AUCを対数変換し、線形混合モデルを当てはめます。固定効果に sequence・period・treatment を置き、順序内の被験者を変量効果(random = ~1 | subject)とすることで、製剤間の比較を被験者内で行います。
# 対数変換した線形混合モデル
fit <- lme(log(AUC) ~ sequence + period + treatment,
random = ~ 1 | subject,
data = dat)
summary(fit)$tTable
Value Std.Error DF t-value p-value
(Intercept) 4.605911 0.05238120 46 87.928449 1.05e-58
sequenceTR -0.018342 0.07221564 46 -0.253989 8.01e-01
period2 0.004517 0.01893210 46 0.238612 8.12e-01
treatmentT -0.038721 0.01893210 46 -2.045282 4.65e-02
注目するのは treatmentT の行です。対数スケールの処置差 \(\hat\delta = -0.0387\)、その標準誤差 \(SE = 0.0189\)、自由度は46です。sequence と period の効果はともに有意でなく(p≈0.80, 0.81)、繰り越し効果や期間効果が結果を歪めていないことを示唆します。treatmentT はわずかに負(Tの方がAUCがやや低い)で、真値 GMR=0.95 を反映しています。
続いて、この処置効果の点推定と90%信頼区間を取り出し、指数変換してGMRと90%信頼区間(パーセント表示)に変換します。BEでは片側5%の2つの検定に対応させるため、信頼水準は90%(level = 0.90)を指定する点に注意します。
# 90%信頼区間を抽出(level=0.90 がBEの肝)
ci <- intervals(fit, level = 0.90, which = "fixed")$fixed
delta_hat <- ci["treatmentT", "est."]
lower_log <- ci["treatmentT", "lower"]
upper_log <- ci["treatmentT", "upper"]
# 指数変換してGMRと90%CI(%表示)へ
GMR <- exp(delta_hat) * 100
CI_low <- exp(lower_log) * 100
CI_high <- exp(upper_log) * 100
round(c(GMR = GMR, CI_lower = CI_low, CI_upper = CI_high), 1)
GMR CI_lower CI_upper
96.2 91.5 101.1
幾何平均比(GMR)は 96.2%、その90%信頼区間は [91.5%, 101.1%] と求まりました。対数スケールの差 −0.0387 を指数変換すると \( \exp(-0.0387) = 0.962 \) となり、GMR 96.2% と整合します。区間が点推定の左右で 91.5〜101.1 とわずかに非対称なのは、対数スケールの対称な区間を指数変換したためです。
最後に、この90%信頼区間が同等域 80〜125% に完全に含まれているかを判定します。
# 同等性の判定
equiv <- (CI_low >= 80) & (CI_high <= 125)
cat("90% CI: [", round(CI_low, 1), ",", round(CI_high, 1), "]\n")
cat("同等域 80-125% に含まれるか:", equiv, "\n")
90% CI: [ 91.5 , 101.1 ]
同等域 80-125% に含まれるか: TRUE
90%信頼区間 [91.5%, 101.1%] は、下限91.5% > 80%、上限101.1% < 125% であり、同等域 [80%, 125%] に完全に含まれています。したがって TOSTの2つの片側検定はともに棄却され、試験製剤Tは標準製剤Rと生物学的に同等であると結論できます。GMRが100%付近にあるだけでなく、区間全体が同等域に収まっている点が決め手です。本来はAUCに加えてCmaxについても同じ手順を踏み、両方が同等域を満たして初めて同等と判定します。
結果を一覧にまとめると、判定の全体像が把握しやすくなります。
| PKパラメータ | GMR | 90%信頼区間 | 同等域 | 判定 |
|---|---|---|---|---|
| AUC | 96.2% | [91.5%, 101.1%] | 80〜125% | 同等 |
なお、この区間の幅は被験者内変動(被験者内CV)の大きさで決まります。被験者内CVが大きい薬剤ほど標準誤差が大きくなり、90%信頼区間が広がって同等域からはみ出しやすくなるため、より多くの被験者数が必要になります。逆に、必要なサンプルサイズは事前に想定する被験者内CVと真のGMRから計算できます。Rでは PowerTOST パッケージの sampleN.TOST() 関数を使うと、目標検出力(通常80〜90%)を満たすために必要な被験者数を簡単に求められます。試験を計画する段階では、解析と並行してこのサンプルサイズ設計を必ず行っておきましょう。
実務でのポイント ― 高変動薬・規制ガイドライン
ここまでで、標準的な2×2クロスオーバー試験における生物学的同等性(BE)判定の枠組みを確認してきました。実務では、薬剤の性質や規制当局ごとの要件によって、追加的な配慮が必要になる場面が少なくありません。ここでは、現場で特に問題になりやすい高変動薬への対応と、主要な規制ガイドラインの考え方を整理します。
高変動薬への対応 ― 同等域の拡大とレプリケートデザイン
被験者内変動係数(within-subject CV)が概ね30%を超える薬剤は「高変動薬(highly variable drug)」と呼ばれます。こうした薬剤では、製剤としての品質に問題がなくても、生理的なばらつきだけで幾何平均比の90%信頼区間が広がりやすく、従来の80〜125%という固定された同等域では同等性を示すことが構造的に困難になります。
この問題に対し、各規制当局は参照製剤の変動の大きさに応じて同等域を調整する 参照スケール平均生物学的同等性(RSABE:Reference-Scaled Average Bioequivalence) の考え方を導入しています。これは、参照製剤(R)の被験者内変動が大きいほど、許容する同等域を統計的に拡大する仕組みです。
RSABEを適用するには、各被験者の参照製剤の変動を直接推定する必要があるため、通常の2期2製剤デザインでは不十分で、レプリケートデザインが用いられます。代表的なものは次の2つです。
| デザイン | 投与パターンの例 | 特徴 |
|---|---|---|
| 部分反復(3期) | TRR / RTR / RRT | 参照製剤Rを2回投与し、Rの被験者内変動のみを推定 |
| 完全反復(4期) | TRTR / RTRT | T・Rとも2回投与し、両製剤の変動を推定可能 |
レプリケートデザインは1人の被験者に同じ製剤を複数回投与するため、washout期間の設計や被験者の負担増、脱落リスクの上昇といった運用面の課題も伴います。高変動薬のBE試験を計画する際は、症例数設計の段階からこれらを織り込んでおくことが重要です。
規制ガイドライン ― 国際的に共通する判定基準
BE試験の判定基準は、国・地域によって細部の運用に差はあるものの、根幹となる考え方は国際的に共通しています。
| 地域・当局 | 主なガイドライン | 基本的な判定基準 |
|---|---|---|
| 日本(厚生労働省) | 後発医薬品の生物学的同等性試験ガイドライン | AUC・Cmaxの幾何平均比の90%信頼区間が80〜125% |
| 米国(FDA) | Bioequivalence Studies に関するガイダンス | 同上(80.00〜125.00%) |
| 欧州(EMA) | Guideline on the Investigation of Bioequivalence | 同上(高変動薬はCmaxの域拡大を許容) |
いずれの当局も、対数変換したAUCとCmaxの幾何平均比について、90%信頼区間が80〜125%に収まることを基本とします。評価指標としては、吸収の総量を表すAUCと、吸収速度を反映するCmaxが中心です。また、食事の影響を受けやすい製剤では空腹時試験に加えて食後試験が求められるなど、製剤特性に応じた試験設計が必要になります。
クロスオーバー試験では、期間1で投与した製剤の影響が期間2に持ち越されないよう、十分な休薬(washout)期間を設ける必要があります。一般に消失半減期の5倍以上が目安とされますが、これが不足するとキャリーオーバー効果が生じ、製剤間の差と期・順序の効果が交絡して、BE評価そのものが成立しなくなる恐れがあります。
クロスオーバー試験では、片方の期で脱落した被験者はその対の情報を活かせず、解析対象から外れて検出力を損ないます。脱落が一定割合生じる前提で症例数を上乗せしておくのが実務的です。また外れ値は安易に除外せず、原因(投与ミス・採血ミス・嘔吐等)を確認したうえで事前規定の手順に従って扱います。Cmaxは単一時点の測定値で変動が大きく、AUCより同等性を示しにくい点にも留意が必要です。
高変動薬ではRSABEとレプリケートデザインで対応し、判定基準(90%CIが80〜125%)は日米欧で共通です。washout・脱落・外れ値・Cmaxの変動という4つの落とし穴を、計画段階のプロトコルと統計解析計画書(SAP)であらかじめ手当てしておくことが、円滑なBE評価の鍵になります。
この記事をより深く理解するための参考書籍
BE試験は、統計学(クロスオーバー解析・信頼区間)と薬物動態学(AUC・Cmax・バイオアベイラビリティ)の両方にまたがるテーマです。それぞれの土台を固めるための書籍を3冊紹介します。






関連記事・次のステップ
BE試験は、クロスオーバーデザイン・同等性評価・症例数設計といった、臨床試験統計の基礎要素が交差するテーマです。各要素をさらに掘り下げたい方は、以下の記事もあわせてご覧ください。
BE試験の土台である2×2クロスオーバーの理論と運用については、クロスオーバー試験の理論と実務で、期・順序効果やキャリーオーバーの扱いをより詳しく解説しています。
「90%信頼区間が同等域に収まる」という考え方は、同等性試験全般に共通する判定の枠組みです。その理論的背景や非劣性との違いは、非劣性試験(Non-inferiority Trial)設計と解析 ― マージン設定で整理しています。
そして、高変動薬で問題になる症例数の確保については、非劣性試験と同等性試験における症例数設計が直接の参考になります。試験デザイン全体を俯瞰したい場合は、臨床試験でよく使われる試験デザイン完全ガイドもご活用ください。
まとめ
本記事では、後発医薬品(ジェネリック)の承認に不可欠な生物学的同等性(BE)試験について、2×2クロスオーバーデザインによるデータ取得から、対数変換・TOST・幾何平均比の90%信頼区間による同等性判定までを通して解説しました。BE試験の本質は、「先発品と後発品で、体内に取り込まれる薬物の量(AUC)と速度(Cmax)が実質的に同じである」ことを、統計的に保証する点にあります。対数変換した指標の幾何平均比の90%信頼区間が80〜125%に収まるかどうか、という基準は、日本・米国・欧州で共通する国際的な合意事項です。
実務では、高変動薬への対応としてRSABEやレプリケートデザインが用いられ、washoutの設計や脱落・外れ値の扱いといった運用上の配慮が判定の信頼性を左右します。これらは統計解析の技術であると同時に、試験計画の段階でプロトコルとSAPに織り込んでおくべき設計思想でもあります。
BE試験が果たす役割は、単なる承認手続きにとどまりません。先発品と治療効果が同等であることを科学的に裏づけることで、ジェネリック医薬品の品質を保証し、患者さんが安心して使える選択肢を増やします。その結果として、医療の質を保ちながら薬剤費を抑制し、持続可能な医療制度を支えるという、公衆衛生上の大きな意義につながっています。一見地味に見えるBE試験が、社会全体の医療を下支えしているといえるでしょう。
BE試験の土台となるクロスオーバーデザインや同等性評価の考え方をさらに深めたい方は、クロスオーバー試験の理論と実務もあわせてお読みいただき、ご自身の研究や業務で確かなBE評価を実現していただければと思います。