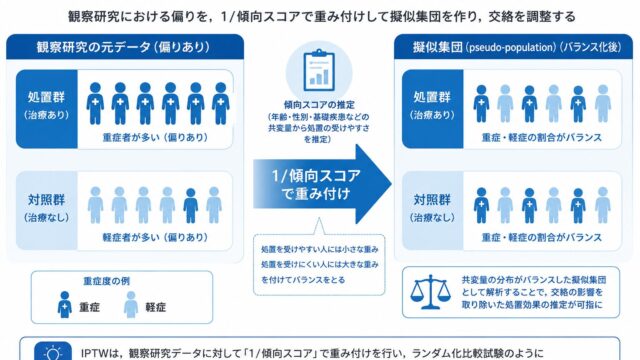
外部対照群(External Control Arm)とは ― FDA・PMDAの規制動向と臨床試験での活用 (1/2) ―

- 外部対照群(External Control Arm, ECA)とは何か、同時対照との違い
- 外部対照群の4つの典型パターン(歴史的/同時期他施設/レジストリ/RWD)
- FDA・EMA・PMDA・ICH E10の最新スタンス
- 希少疾患・がん領域で外部対照群が広がる構造的な理由
- 運用上の限界と、計画段階で必ず検討すべき7つの注意点
はじめに:なぜ今「外部対照群」を学ぶ必要があるのか
製薬開発における無作為化二重盲検試験(RCT)が、承認エビデンスのゴールドスタンダードであることは今も変わりません。しかし、希少疾患・小児疾患・進行がん・致死的疾患など、倫理的・実務的にプラセボ群や標準治療群を組めない場面が急速に増えています。患者数が極端に少ない領域では、対照群への組み入れだけで承認申請のスケジュールが数年単位で後ろ倒しになることも珍しくありません。
そこで近年、急速に注目を集めているのが 外部対照群(External Control Arm, 以下 ECA) です。被験薬群は通常通り組み入れる一方で、対照群を「試験外」のデータソース(過去試験・レジストリ・電子カルテなど)から別途構築するアプローチで、FDAは2023年2月に専用のドラフトガイダンス “Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products” を発出しました。PMDAも「リアルワールドデータ(RWD)の利活用」の文脈で、レジストリ由来の外部対照を具体的に整理しつつあります。
本記事は2部構成の前編として、ECAの全体像・パターン分類・規制当局のスタンスを整理します。続編では統計手法(傾向スコアマッチング・MAIC・ベイズ動的借用)の実装に踏み込みます。
外部対照群とは ― 同時対照との違い
外部対照群とは、被験薬群の比較対象を「同じプロトコルで同時期に組み入れた群」ではなく、試験外のデータソースから別途構築した群で代用するデザインです。比較相手が試験プロトコルの外側にいる、というのが定義上の最大のポイントになります。
ICH E10「Choice of Control Group and Related Issues in Clinical Trials」(Step 4: 2000年7月)では、対照群を5タイプに分類しています。外部対照(external / historical control)は5番目に位置づけられ、「比較可能性の確保とバイアス制御に深刻な懸念があるため、特殊な状況でのみ使用可能」と明示されています。20年以上前のこの位置づけが、現在のECA運用の出発点であり、規制当局が一貫して慎重姿勢を保つ根拠でもあります。
| タイプ | 対照の種類 | 特徴 |
|---|---|---|
| ① | プラセボ対照 | 同時並行・盲検性確保が容易 |
| ② | 無治療対照 | 同時並行・盲検性は限定的 |
| ③ | 用量対照 | 同時並行・用量反応評価に有用 |
| ④ | 実薬対照 | 同時並行・非劣性/優越性試験 |
| ⑤ | 外部対照(歴史的対照) | 非同時並行・特殊な状況のみ |
ICH E10が示した「特殊な状況」とは、概ね次のような場面です:
- 病気の自然経過が極めて予測可能(治療しなければ確実に死亡または進行する)
- 治療効果が劇的(dramatic effect)で、過去データとの比較でも誤りにくい
- ランダム化が倫理的に困難(小児希少疾患・致死的疾患・標準治療がない領域)
逆に、効果がプラセボとの差で5〜10%程度しかない領域では、外部対照はほぼ常に不適切になります。「効くか効かないか微妙な薬」を外部対照で評価しようとしても、バイアスのほうが効果サイズを上回ってしまうためです。一例として、降圧薬や脂質異常症治療薬のように既存薬との小さな差を示すタイプの開発では、過去データとの単純比較で「優れている」ように見えても、それは時代差や患者プロファイルの違いを反映しているだけ、ということが頻繁に起こります。
このため、ECAを採用するかどうかの最初の判断は、統計的な技巧の前に 「対象疾患の自然経過がそもそも外部データで再現できるか」 を見極めることから始まります。ここでの判断を誤ると、後段でどれだけ高度な統計手法を積み上げても、規制当局の評価には耐えられません。
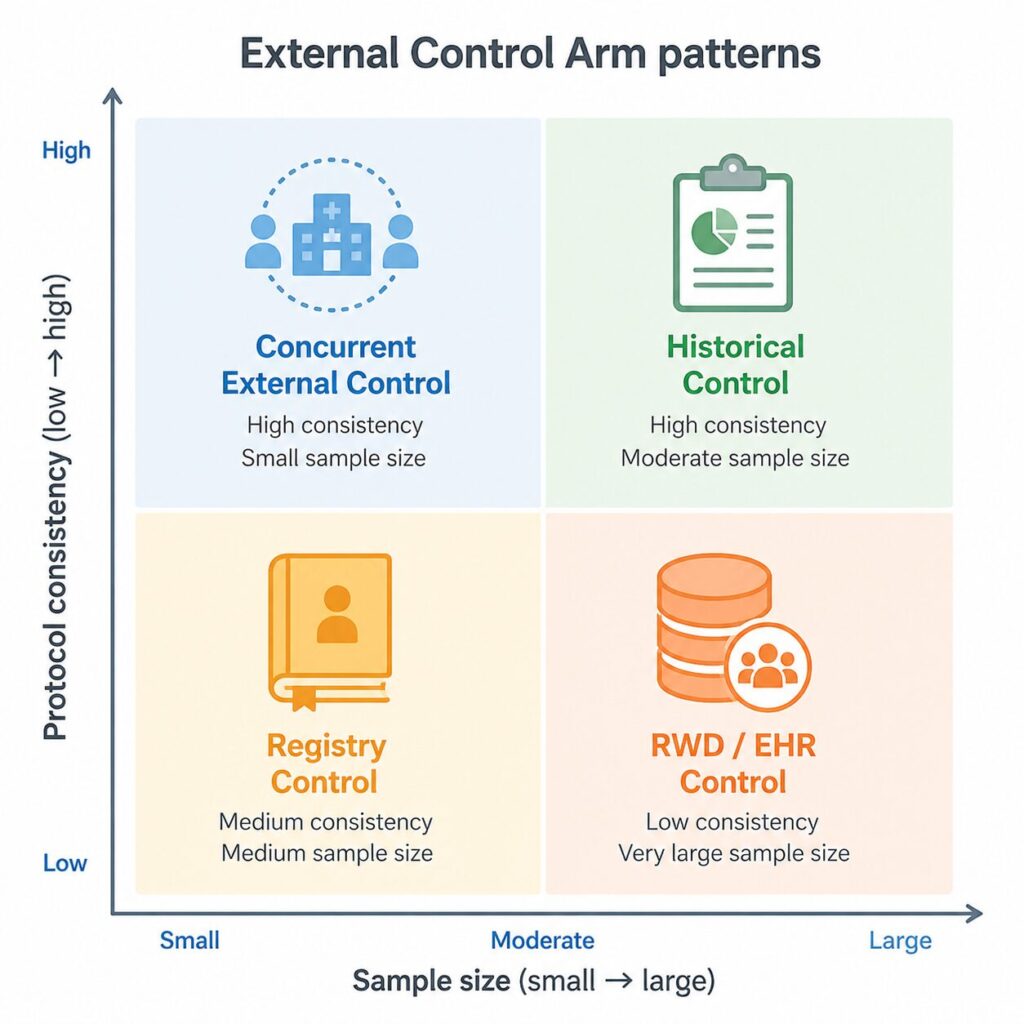
外部対照群の4つの典型パターン
実務で扱う外部対照は、データソースの性質によって大きく4パターンに分かれます。プロトコル整合性とサンプルサイズのトレードオフが、それぞれの強み・弱みを規定しています。
| パターン | データソース | 強み | 主な弱点 |
|---|---|---|---|
| ① 歴史的対照 | 過去の同種試験データ | プロトコル・評価項目が揃いやすい | 時代差・治療標準の進歩 |
| ② 同時期他施設対照 | 並行実施の他試験 | 時代差を緩和 | 施設選択バイアス |
| ③ レジストリ対照 | 疾患レジストリ・自然歴データ | 疾患特異的な経過情報が豊富 | 評価方法のばらつき |
| ④ RWD対照 | 電子カルテ・保険レセプト | サンプルが大規模 | エンドポイント欠落・コーディング誤差 |
近年のFDA承認では ① + ③ または ① + ④ を組み合わせたハイブリッド設計が主流になりつつあります。歴史的試験の正確なエンドポイント情報と、RWD・レジストリの大規模サンプルを組み合わせることで、両者の弱点を相互に補完する狙いです。たとえば、希少がん領域では「過去の単群第II相試験」を主たる外部対照とし、それでは年齢分布や治療歴のバランスが取れない場合に「米国の腫瘍領域RWDから条件マッチで補完」するというハイブリッド構成がよく見られます。
どのパターンを採るにせよ、被験薬群と外部対照群の 共変量分布(年齢・PS・治療歴・バイオマーカーなど)が比較可能であること を、解析計画段階で数値的に示せるかどうかが、規制当局との合意可能性を大きく左右します。
規制当局の見解(FDA・EMA・PMDA・ICH)
FDA:2023年2月のドラフトガイダンス
FDAは2023年2月、“Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products” ドラフトガイダンスを公表し、5月2日までパブリックコメントを募集しました。主な論点は以下の通りです:
- 外部対照のデータソース選定基準(時代・地理・治療標準の整合性)
- バイアス源の系統的評価(選択・測定・交絡・欠測)
- 患者レベルデータをFDAに提供できる契約構造(データオーナーが第三者の場合)
- 申請前の Type C ミーティングで規制側と方針を合意することの推奨
EMA:限定的な肯定姿勢
EMAは早くから希少疾患領域でECAを認めてきましたが、評価軸を 「事前にFDA/EMAと合意していること」「自然歴が確立していること」 に明確に置いています。事後的に過去データを当てる post-hoc 型の外部対照分析には、一貫して厳しい立場です。
PMDA:レジストリ活用を中心に整理中
PMDAは「RWD WG(リアルワールドデータ ワーキンググループ)」を中心に、レジストリの信頼性水準・対面助言での事前合意の重要性を整理しています。希少疾患領域では、自然歴レジストリを外部対照として活用することが具体的に許容されており、対面助言での事前相談が事実上の必須プロセスです。
ICH E10:5番目の対照グループとして位置づけ
ICH E10は2000年策定と古いものの、「特殊な状況(劇的効果+自然経過の予測可能性)」でのみ外部対照を許容する原則は今も有効です。各極の最新ガイダンスはE10の上に積み上がる形で運用されており、まずE10を読むことがECA設計の出発点になります。
規制当局はいずれも 「事前合意(pre-specification)」 と 「バイアスの系統的検討」 を最重要視しています。後出しの外部対照分析は申請段階で却下されやすいため、計画段階から規制側との合意プロセスを必ず組み込むことが必須です。
RWD・外部対照群の設計実務をさらに体系的に学びたい方は、本記事末尾で紹介する『超入門!スラスラわかる リアルワールドデータで臨床研究 第2版』(康永秀生・金芳堂)が最適です。データベース構築から因果推論の入門までを1冊でカバーしています。
なぜ希少疾患・がん領域で広がるのか
ECAが急速に広がっている背景には、3つの構造的な要因があります。
(1) 患者数の絶対的少なさ:希少疾患では対象患者が世界全体で数百〜数千人しかいません。RCTで対照群に半数を割り振ると、検出力を確保するための組み入れに 10年以上かかることも珍しくなく、ビジネス上もアンメットニーズ充足の観点でも現実的ではありません。
(2) プラセボ投与の倫理的困難:進行がん・致死的小児疾患では、プラセボ群への割付が 倫理委員会で承認されないケースが増えています。標準治療がない、または既存治療が無効な領域では、すべての患者に被験薬を提供するシングルアーム設計が望まれます。
(3) RWDデータ基盤の急速な整備:電子カルテ・保険レセプト・疾患レジストリの整備が進み、過去患者の予後データを系統的に取り出せる環境が整いつつあります。Flatiron Health(米国の腫瘍領域RWDベンダー)や IQVIA Oncology のRWDなど、専門ベンダーのデータが申請に使われる事例も増加しています。
実際、近年のFDA承認では、白血病治療薬・希少がん治療薬・小児難病治療薬など、外部対照を活用した申請が複数承認されています。ECAは「例外的な手段」から「条件付きで標準的な選択肢の一つ」へと位置づけが変化しつつあると言えるでしょう。製薬企業の生物統計担当者にとっても、ECAの設計・評価スキルは「特殊な状況のためのオプション」ではなく、開発戦略を組み立てる上で標準的に押さえておくべき領域に変わってきています。
一方で、ECAが向く領域には明確な共通点があります。(a) 自然経過がよく分かっている、(b) エンドポイントが客観的で測定誤差が小さい(全生存期間・無増悪生存期間など)、(c) 治療標準が比較的安定している、の3条件が揃う領域では成功事例が積み上がりやすく、逆に主観的評価指標(PRO・QOLなど)が主要評価項目の場合は外部対照との比較可能性が大きく崩れがちです。設計段階でのエンドポイント選択は、ECAの成否を左右する最初の分岐点です。
外部対照群の限界と注意点(7つのリスク)
ECAは万能ではありません。以下の7点は、計画段階で必ず検討すべき項目です。
| # | リスク | 典型的な対策 |
|---|---|---|
| 1 | 時代バイアス(治療標準の進歩) | 過去データの取得時期を直近5〜10年に限定 |
| 2 | 施設選択バイアス | データソースを複数施設・複数地域に分散 |
| 3 | エンドポイント定義の不一致 | 評価項目の操作的定義を事前文書化 |
| 4 | 欠測データ | 多重代入法・感度分析を計画書に明記 |
| 5 | 未測定交絡 | E-value 等の感度分析を併用 |
| 6 | 治療標準の変化 | 同時期サブグループでバランスチェック |
| 7 | 規制側の事後評価リスク | Pre-specification と FDA/PMDA との事前合意 |
特に 未測定交絡 は外部対照群の最大の弱点です。RCTでは無作為化により未測定交絡が確率的にバランスしますが、外部対照群ではそのような数学的保証はありません。「測れていない要因が結果を変えている可能性」を常に検討し、感度分析(E-value など)で頑健性を示すことが極めて重要になります。
まとめと次回予告
本記事では、外部対照群(ECA)の基本概念・4つの典型パターン・FDA/EMA/PMDA/ICH E10の規制スタンス・運用上の限界点を整理しました。ECAは「特殊な状況でのみ使う例外的デザイン」から、「希少疾患・がん領域における条件付きの標準的選択肢」へと、確実に位置づけが変化しています。
- ECAは「特殊な状況(希少疾患・劇的効果・自然歴予測可能)」で例外的に使用するデザインで、RCTを完全に置き換えるものではない
- FDAは2023年ドラフトガイダンス、PMDAはレジストリ活用、EMAは事前合意重視と、各極とも 事前計画とバイアス系統評価 を最重要視
- 4つのパターン(歴史的/同時期他施設/レジストリ/RWD)の使い分けと、ハイブリッド設計が現在の主流
- 未測定交絡が最大の弱点。感度分析と pre-specification で頑健性を担保する
次回予告:続編「外部対照群の統計手法 (2/2)」では、傾向スコアマッチング(PSM)・MAIC(Matching-Adjusted Indirect Comparison)・ベイズ動的借用(dynamic borrowing)の3つの主要手法を、Rコード付きで実装します。未測定交絡への感度分析(E-value)まで踏み込み、実務でそのまま使える内容にする予定です。
関連記事・次のステップ
- ベイズ臨床試験デザイン入門 ― FDA 2026年ドラフトガイダンスから読み解く事前分布・予測確率・適応的デザイン ― — 外部対照群と相性のよいベイズ動的借用の前提知識として
- ICH E20(臨床試験のためのアダプティブデザイン)とは ― Step 4最終化目前!国際調和ガイドラインの全体像 ― — 試験中に外部対照を組み込む適応的設計の枠組み
- ICH E9(R1) Estimandフレームワーク徹底解説 ― 5属性とintercurrent event戦略の使い分け ― — 外部対照を含めた比較における推定対象の整理に
📚 参考書籍