説明可能AI(XAI)とは ― 製薬業界での活用・メリットと規制対応を生物統計家の視点で解説 ―

この記事を読むと、次のことがわかります。
- 製薬業界でAIが、創薬から臨床試験、市販後の安全性監視まで、どの工程でどう使われているか
- 説明可能AI(XAI:人間が予測の根拠を理解できるように工夫されたAI)とは何か、そしてなぜ規制産業である製薬で特に重要なのか
- AIを活用することのメリットと、見落としてはならないデメリット(リスク)
- FDA(米国)・EMA(欧州)・日本(PMDA等)の規制動向と、その中で生物統計家がどう向き合うべきか
- 「予測の当たりやすさ」だけでなく「説明できること」を重視する、生物統計家ならではのバランス感覚
はじめに:なぜ今、製薬業界で「説明できるAI」が問われるのか
新しい医薬品を1つ世に送り出すには、一般に10年を超える歳月と巨額の費用がかかり、しかも開発の成功率は決して高くありません。この厳しい現実を前に、AI(人工知能)によって開発の時間とコストを圧縮できるのではないかという期待が、近年急速に高まっています。
背景にあるのは、扱うデータの爆発的な増大です。臨床試験データに加えて、リアルワールドデータ(RWD:日常診療や保険請求などから得られる実臨床のデータ)、ウェアラブル端末の測定値、医用画像、ゲノム情報など、人手だけで処理するには限界のある大規模・多様なデータが日々生まれています。さらに、ChatGPTやClaudeに代表される生成AIの普及により、文書作成やプログラム生成といった実務での利用が一気に現実のものとなりました。
しかし、製薬は厳格に規制された産業です。AIが「なぜその予測に至ったのか」を説明できないブラックボックス問題は、規制当局への説明責任が求められる場面で大きな障壁になります。だからこそ今、「説明できるAI」=説明可能AI(XAI)が問われているのです。
本記事は、製薬企業で統計解析やデータサイエンスに携わる方、そしてこれから統計を学ぶ学生の方を念頭に、AIの活用とXAIをめぐる論点を生物統計家の視点から整理していきます。
製薬業界でAIはどこまで使われているか ― 創薬から市販後まで
医薬品が患者さんの手元に届くまでには、創薬から市販後の安全性監視に至る長いバリューチェーン(価値連鎖)があります。AIは、その各段階に少しずつ浸透しつつあります。まずは全体像を整理してみましょう。
| 開発段階 | 主なAI活用例 | 期待される効果 |
|---|---|---|
| 創薬(ディスカバリー) | 候補化合物・創薬標的の探索、化合物の特性予測、膨大な探索空間の絞り込み | 有望な候補へ早く到達し、試行錯誤を削減 |
| 臨床試験 | 患者リクルートの最適化、実施医療機関(サイト)選定、試験デザイン支援、デジタルツインや外部対照(合成対照群)の構築、画像・ウェアラブルからのエンドポイント自動評価 | 組入れの効率化と試験運営の最適化 |
| データ解析・統計 | 大規模データからのパターン抽出、サブグループ探索、予測モデルの構築 | 仮説生成と探索的解析の高速化 |
| ファーマコビジランス(市販後の安全性監視) | 有害事象・安全性シグナルの自動検出、症例報告の処理効率化 | 安全性情報の早期把握と業務負荷の軽減 |
| 製造・品質(CMC) | 工程管理、品質予測、逸脱検知 | 品質の安定化と不良の未然防止 |
| 文書作成・業務支援 | CSR(治験総括報告書)や申請資料のドラフト、SAS/Rプログラムの生成補助、照会事項対応の下書き | 定型業務の省力化(要・人間の検証) |
創薬(ディスカバリー)の段階では、無数にある候補化合物や創薬標的の中から有望なものを探し当て、化合物の特性を予測して探索空間を絞り込む用途でAIが使われています。続く臨床試験では、患者リクルートの最適化や実施医療機関(サイト)の選定、試験デザインの支援に加え、デジタルツインや外部対照(合成対照群:過去データなどから構成する比較対照)の構築、画像やウェアラブルからエンドポイント(評価項目)を自動的に測定する試みも進んでいます。
データ解析・統計の領域では、大規模データからのパターン抽出やサブグループ探索、予測モデルの構築にAIが力を発揮します。ただし注意したいのは、承認申請の根拠となる確証的解析の主役は、依然として事前に規定された統計手法だという点です。AIが得意とするのはあくまで探索や補助であり、検証すべき「効果」をどう定義し、どう推定するかという土台が揺らいではいけません。解析でAIが扱う「効果」の定義を厳密にする考え方については、Estimandを理解するためにもあわせてご覧ください。
市販後に目を移すと、ファーマコビジランス(市販後の安全性監視)では有害事象や安全性シグナルの自動検出、症例報告の処理効率化が期待されています。製造・品質(CMC)の分野でも、工程管理・品質予測・逸脱検知にAIが活用されつつあります。そして、生成AIがとりわけ得意とするのが文書作成・業務支援です。CSR(治験総括報告書)や申請資料のドラフト、SAS/Rプログラムの生成補助、照会事項対応の下書きなど活躍の場は広いものの、最終的には必ず人間による検証が欠かせません。
このようにAIは創薬から市販後まで幅広く浸透していますが、いずれの場面でも「予測の根拠が不透明なまま」では、規制当局に対する説明責任を果たせません。なぜその結論に至ったのかを示せること ── それこそが、次章で扱う説明可能AI(XAI)が求められる理由です。
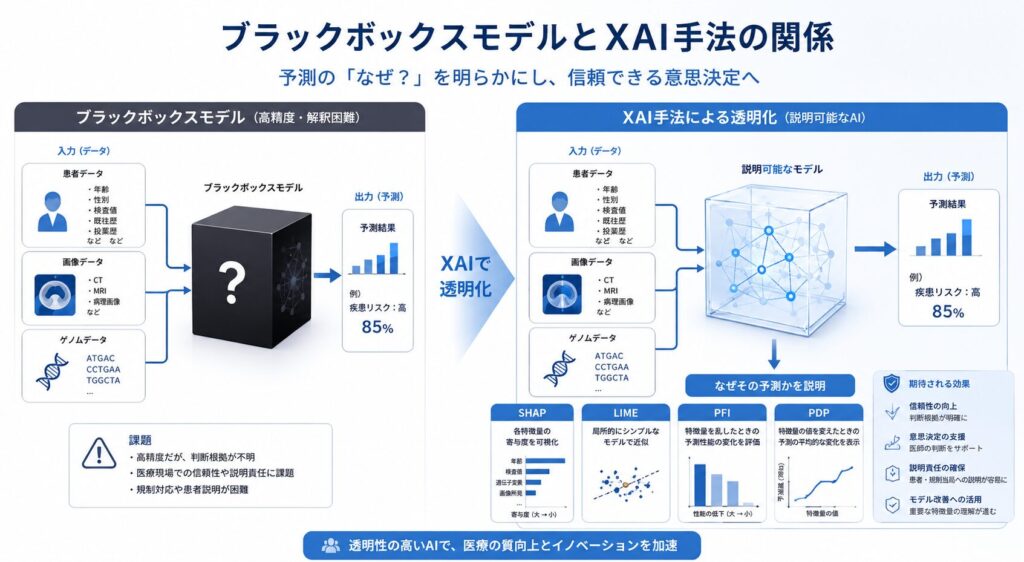
説明可能AI(XAI)とは ― ブラックボックス問題を生物統計家の視点で読み解く
説明可能AI(XAI:eXplainable AI)とは、ブラックボックスなAIモデルの予測根拠を、人間が解釈・検証できるようにする手法群の総称です。ここでいう「人間」とは、モデルを作る開発者だけではありません。承認の可否を判断する規制当局、目の前の患者に治療方針を説明する医師、そして自らの治療を理解したい患者まで、すべてが対象に含まれます。
なぜいま、説明可能性がこれほど重視されるのでしょうか。背景にあるのが、いわゆる「ブラックボックス問題」です。ディープラーニング(深層学習)、勾配ブースティング木(GBDT:複数の決定木を順に組み合わせる手法)、ランダムフォレストといった現代のモデルは非常に高い予測精度を誇りますが、「なぜその予測になったのか」という根拠が極めて不透明です。一般に、予測精度と解釈性(なぜそう予測したかの分かりやすさ)はトレードオフの関係にあり、精度を追えば追うほど中身が見えにくくなりがちです。
医療・製薬のような規制産業では、この「説明できなさ」が決定的な障壁になります。どれほど精度が高くても、根拠を示せないモデルは「信頼」を獲得できず、ひいては「規制承認」のハードルを越えられないからです。そこで登場するのがXAIの各種手法です。代表的なものを整理してみましょう。
| 手法 | 考え方 | 特徴・使いどころ |
|---|---|---|
| SHAP (SHapley Additive exPlanations) | 協力ゲーム理論の「Shapley値」に基づき、各特徴量が予測値にどれだけ寄与したかを公平に分配・定量化する。 | XAIで最もよく使われる定番手法。個々の予測(局所)から全体傾向(大域)まで一貫して説明できる。 |
| LIME (Local Interpretable Model-agnostic Explanations) | 個々の予測の周辺で、単純で解釈可能なモデルを使って局所的に近似し説明する。 | 「この症例がこう判定された理由」を1件ずつ示したいときに有効。局所説明に特化。 |
| PFI (Permutation Feature Importance) | ある特徴量の値をシャッフルし、予測性能がどれだけ劣化するかを見ることで重要度を測る。 | モデル全体でどの変数が効いているか(大域的な重要度)を手早く把握したいときに便利。 |
| PDP / ICE (Partial Dependence Plot / Individual Conditional Expectation) | 特徴量の値と予測との関係をグラフで可視化する。 | 「用量が増えると予測リスクはどう動くか」など、変数と予測の関係性を直感的に示したいときに使う。 |
ここに挙げた4手法は、いずれもモデル非依存(model-agnostic)、すなわち中身がどんなアルゴリズムであっても外側から適用できる点が共通しています。なかでもSHAPの土台となるShapley値は、もともと協力ゲーム理論に由来する数理で、ここに生物統計家ならではの視点が効いてきます。
第一に、SHAPの理論的支柱であるShapley値は協力ゲーム理論に由来し、統計家・計量経済学者にとって馴染みのある数理です。各プレイヤー(特徴量)の貢献を公平に分配するという発想は、分散の寄与分解などと地続きであり、生物統計家はXAIの理論的背景を理解しやすい立場にあります。
第二に、より本質的な注意点として、「特徴量の重要度・寄与」は関連(association)であって因果効果(介入効果)ではないという点です。SHAP値が大きいからといって、それが「原因」だとは限りません。交絡(本当の原因が別にある状態)や選択バイアスが背後にあれば、寄与の大きさは容易にミスリードを生みます。こうした落とし穴を見抜く統計的素養こそが、XAIの誤読を防ぐ最大の武器になります。
最後に、説明可能性へのアプローチには大きく2つの方向性があることも押さえておきましょう。1つは、ロジスティック回帰・一般化加法モデル(GAM)・決定木のように最初から解釈可能なモデルを使う設計(interpretable-by-design)です。もう1つは、高精度なブラックボックスをまず構築し、SHAPやLIMEで事後的に説明を付与する(post-hoc XAI)アプローチです。精度を優先するなら後者、説明責任を最優先するなら前者というように、用途と規制要件に応じた使い分けが求められます。
製薬業務でAIを活用するメリットとデメリット
それでは、製薬業務にAIを取り入れることで、具体的に何が得られ、何に注意すべきなのでしょうか。メリットとデメリットを観点ごとに対比して整理します。
| 観点 | メリット | デメリット・注意点 |
|---|---|---|
| 開発スピード・コスト | 候補化合物の探索、文書ドラフト作成、コード生成補助などで開発を高速化・低コスト化できる。 | 生成AIの出力にはハルシネーション(事実でない内容の生成)が混在し、検証なしの利用は危険。 |
| データ活用 | 画像・ゲノム・ウェアラブル・RWD(リアルワールドデータ)など大規模・多様なデータからパターンを抽出できる。 | データのバイアスや代表性不足があると、小集団で性能が劣化し健康格差を拡大しうる。 |
| 臨床試験の効率 | 患者リクルートやサイト(実施施設)選定の最適化により、試験運営を効率化できる。 | データドリフトや過学習により、環境変化に伴ってモデル性能が経時的に劣化しうる。 |
| 安全性監視 | ファーマコビジランス(市販後の安全性監視)で、安全性シグナルの検出を効率化できる。 | ブラックボックス性のため、規制当局・医師・患者への説明責任を果たしにくい。 |
| 業務効率全般 | ヒューマンエラーや単純作業を削減し、人的リソースを高度な判断に振り向けられる。 | 再現性・監査可能性・バリデーション負荷、データプライバシー・知的財産・責任の所在が課題となる。 |
メリットの多くは「効率化」と「これまで扱えなかったデータの活用」に集約されますが、生物統計家として特に目を光らせたいのは、表の右列に並んだリスクです。なかでも次の3つは、統計的に深刻な落とし穴を含んでいます。
① データのバイアス・代表性不足:学習データが特定の集団に偏っていると、小児・希少疾患・特定人種といった小集団でモデル性能が大きく劣化します。これは単なる精度の問題にとどまらず、健康格差を拡大しかねない倫理的リスクです。
② データドリフト/過学習:診療環境や患者集団は時間とともに変化します。開発時に高精度でも、環境変化によってモデル性能が経時的に劣化(ドリフト)し、過学習していれば本番データで通用しません。継続的なモニタリングが不可欠です。
③ ハルシネーション:生成AIは、もっともらしく見える事実でない内容を生成することがあります。文書ドラフトや要約に用いる場合でも、人間による検証を欠いた利用は重大な誤りにつながります。
こうしたリスクは、いずれも「モデルが何を根拠に、どう振る舞っているか」を見えるようにしなければ管理できません。だからこそ、本記事の冒頭から述べてきた説明可能性(XAI)と、それを担保する規制対応が決定的に重要になります。次章では、こうしたリスクに製薬業界がどう向き合い、規制当局がどのような枠組みを示しているのかを見ていきます。
規制はどう動いているか ― FDA・EMA・日本の最新動向
製薬業界でAI、とりわけ説明可能AI(XAI)を申請業務に使おうとすると、まず気になるのが「当局はこれをどう見ているのか」という点です。ここでは米国・欧州・日本の3極について、2025〜2026年時点の最新動向を整理します。
まず米国のFDA(食品医薬品局)です。2025年1月、FDAは医薬品開発分野で初の本格的なAIガイダンス案「Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products(規制上の意思決定を支援するためのAI利用に関する考慮事項)」を公表しました。適用範囲は広く、非臨床・臨床・市販後・製造の各段階で、AIが安全性・有効性・品質といった規制上の意思決定を支える情報やデータを生み出す用途を対象とします。
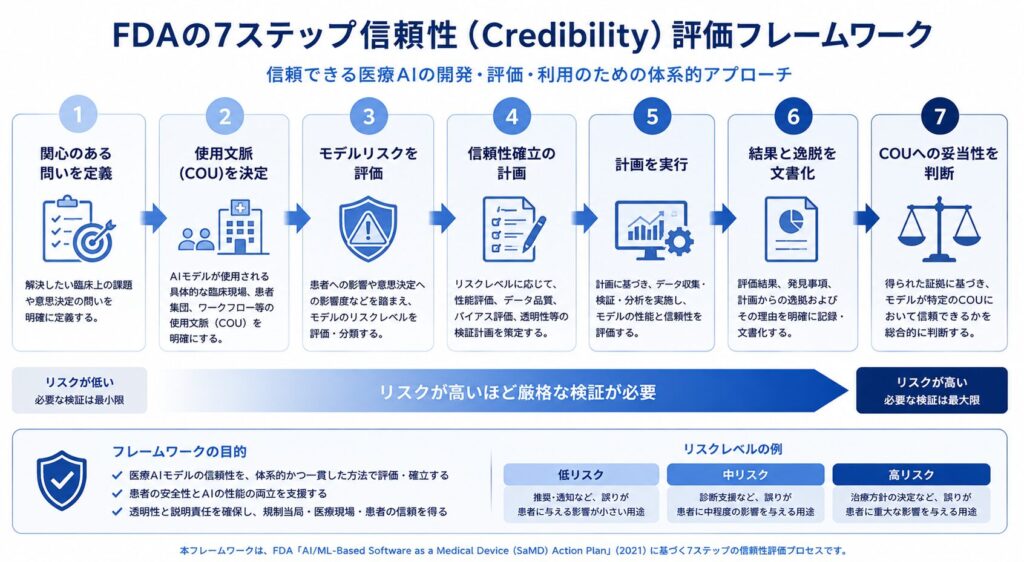
このガイダンス案の中核にあるのが、リスクベースの「信頼性(credibility)評価フレームワーク」です。ここでいう信頼性とは、特定の使用文脈(Context of Use, COU)におけるモデル性能への信頼を指します。同じAIモデルでも「どの場面で、何を判断するために使うのか」によって求められる証拠の重さが変わる、という考え方です。FDAはこの信頼性を確立するための手順を、次の7ステップとして示しています。
1) 関心のある問い(question of interest)を定義する
2) AIモデルのCOU(使用文脈)を決定する
3) AIモデルのリスクを評価する(モデルの影響度 × 誤った場合の結果の重大性)
4) 信頼性を確立する計画を立てる
5) 計画を実行する
6) 結果と計画からの逸脱を文書化する
7) COUに照らしてモデルの妥当性を判断する
ポイントは、リスクが高いほど(重要な意思決定へのAIの影響が大きいほど)、より厳格な証拠が求められるという点です。なお、このガイダンス案のパブリックコメントは2025年4月7日に締め切られ、最終化はおおむね2026年(2026年第2四半期)に見込まれており、現在はまさに最終化の段階にあります。申請データの取り扱いという観点では、こうしたAI利用の文書化は、従来の申請電子データ提出についての延長線上で考えると理解しやすいでしょう。
次に欧州のEMA(欧州医薬品庁)です。EMAは2024年9月9日、AIの医薬品ライフサイクル利用に関する最終リフレクションペーパー(文書番号 EMA/CHMP/CVMP/83833/2023、ドラフトは2023年7月公表)を採択しました。こちらもリスクベースですが、特徴は「人間中心(human-centric)」アプローチを掲げている点です。対象は創薬・臨床試験・ファーマコビジランス(市販後の安全性監視)まで全ライフサイクルに及びます。データの品質と代表性を重視し、小児や希少疾患といった小集団でのバイアスに注意を促すとともに、最終的な責任は人間、すなわちマーケティング承認取得者(MAH)にあると明記しています。
最後に日本です。2025年には日本初のAIに関する基本法である「AI推進法」が成立し、リスクベースの「AI事業者ガイドライン」も整備されています。医療AIの多くはプログラム医療機器(SaMD)として薬機法で規制され、PMDA(医薬品医療機器総合機構)はSaMDとリアルワールドデータ(RWD)に関するシンポジウムを2026年2〜3月に開催しました。2026年度の診療報酬改定でもAI・ICT活用の促進が盛り込まれています。ただし、創薬や統計解析そのものへのAI利用に特化した日本独自の規制は、診断支援を担う医療機器分野が先行しており、欧米に比べるとなお整備・検討が進む途上の段階にあるといえます。
3極の動向を整理すると、次のようになります。
| 地域/当局 | 主要文書・枠組み | 特徴・ポイント |
|---|---|---|
| 米国/FDA | AI利用ガイダンス案(2025年1月公表、最終化見込み) | リスクベースの信頼性(credibility)評価フレームワーク。COU(使用文脈)を起点とする7ステップ。高リスク用途ほど厳格な証拠を要求。 |
| 欧州/EMA | 最終リフレクションペーパー(EMA/CHMP/CVMP/83833/2023、2024年9月採択) | リスクベースかつ人間中心(human-centric)。全ライフサイクル対象。データの代表性・小集団バイアスを重視し、最終責任はMAH(人間)に。 |
| 日本/PMDA・厚労省 | AI推進法(2025年成立)、AI事業者ガイドライン、薬機法(SaMD) | 医療機器(診断支援)分野が先行。創薬・統計解析へのAI利用に特化した規制は整備途上。 |
製薬分野におけるAI規制は、現在まさに形成されつつある発展途上の領域です。FDAガイダンスの最終版や各当局の文書は今後更新される可能性が高く、本記事の内容は執筆時点の情報に基づくものです。実際の申請や開発判断にあたっては、必ずFDA・EMA・PMDAなど各当局が公表する最新の一次文書を確認してください。
生物統計家はAI時代にどう向き合うか ― 実務のポイント
ここまで3極の規制動向を見てきて、お気づきの方も多いと思います。FDAの「まずCOU(使用文脈)を定義する」という発想は、私たち生物統計家が当たり前に行ってきた仕事の進め方とほとんど同じなのです。
統計解析計画書(SAP, Statistical Analysis Plan)を解析の前にきちんと定め、「何を、どの集団で、どう評価するのか」を事前に規定する。この事前規定(pre-specification)の文化こそ、AIの信頼性評価フレームワークが求めているものの本質です。使用文脈を最初に言語化し、リスクの高さに応じて検証の深さを決め、手順と逸脱を徹底的に文書化する――これは生物統計家が最も得意とする領域だといえます。
実務上の勘所をいくつか挙げておきます。第一に、解釈可能性を最初から設計に織り込む(interpretable-by-design)か、ブラックボックスなモデルを使うなら事後的な説明(XAI)とバリデーションを必ずセットにすることです。第二に、AIの出力を鵜呑みにせず、交絡や選択バイアス、データの代表性を批判的に評価し、相関と因果をきちんと区別することです。因果推論の素養を持つ生物統計家だからこそ果たせる役割がここにあります。
そして最も重要なのが、Human-in-the-loop、すなわち最終的な判断と説明責任は人間が担保するという原則です。これはFDAの7ステップにもEMAの人間中心アプローチにも共通する考え方です。生成AIによるコード生成や文書のドラフト作成は、あくまで作業を効率化する「補助」にすぎません。その出力を検証し、責任を持って世に出すのは私たち人間の仕事です。
生物統計家が長年培ってきた「事前規定(pre-specification)」「リスクベースの検証」「交絡と因果の峻別」という素養は、AI時代にこそ価値を持ちます。使用文脈の定義から検証、説明責任の担保まで、AIガイダンスが求める要件の多くは生物統計の発想と地続きです。AIを脅威ではなく強力なツールとして使いこなし、その出力に責任を持って意味づけを与える――それがこれからの生物統計家に求められる役割です。
📚 この記事をより深く理解するための参考書籍
説明可能AI(XAI)と、それを支える統計・機械学習の素養をさらに深めたい方に、おすすめの書籍を3冊ご紹介します。






関連記事・次のステップ
AIと規制・統計解析のつながりをさらに深めたい方は、あわせて以下の記事もご覧ください。
- 申請電子データ提出について ― AI利用の文書化とも地続きの、規制当局への電子データ提出の基礎を解説しています。
- Estimandを理解するために ― 「何の効果を推定したいのか」を厳密に定義するEstimandの考え方は、AI時代の解析設計の出発点になります。
- 一般化推定方程式(GEE)を徹底解説 ― 解釈可能な統計モデルの代表例として、相関構造を扱うGEEの数理を学べます。
まとめ
本記事では、製薬業界における説明可能AI(XAI)の活用と規制対応を、生物統計家の視点から整理しました。AIは創薬から臨床試験、データ解析、ファーマコビジランス、製造、文書作成まで幅広く浸透しつつあり、開発の高速化やこれまで扱えなかったデータの活用といった大きなメリットをもたらします。その一方で、ブラックボックス性・データのバイアス・データドリフト・ハルシネーションといったリスクは、統計的にも倫理的にも軽視できません。
こうしたリスクに向き合う鍵が、予測の根拠を人間が理解できるようにする説明可能AI(XAI)です。SHAPをはじめとする手法はモデルの中身を可視化してくれますが、「寄与は因果ではない」という統計家の批判的視点があってはじめて正しく読み解けます。FDAの信頼性評価フレームワーク、EMAの人間中心アプローチ、そして日本の整備途上の枠組みは、いずれも「使用文脈の定義」「リスクベースの検証」「人間による最終責任」という共通点を持ち、これらは生物統計家が長年培ってきた事前規定と検証の文化と見事に重なります。
AIを脅威としてではなく、責任を持って意味づけを与えるべき強力なツールとして使いこなすこと。それこそが、これからの製薬業界で生物統計家が発揮できる最大の強みになります。