FDA Real-World Evidence (RWE) ガイダンス総まとめ ― 8つの公式文書で読み解く規制活用の全体像 ―

- FDA Real-World Evidence (RWE) プログラムの法的根拠(21st Century Cures Act §3022・FD&C Act §505F)
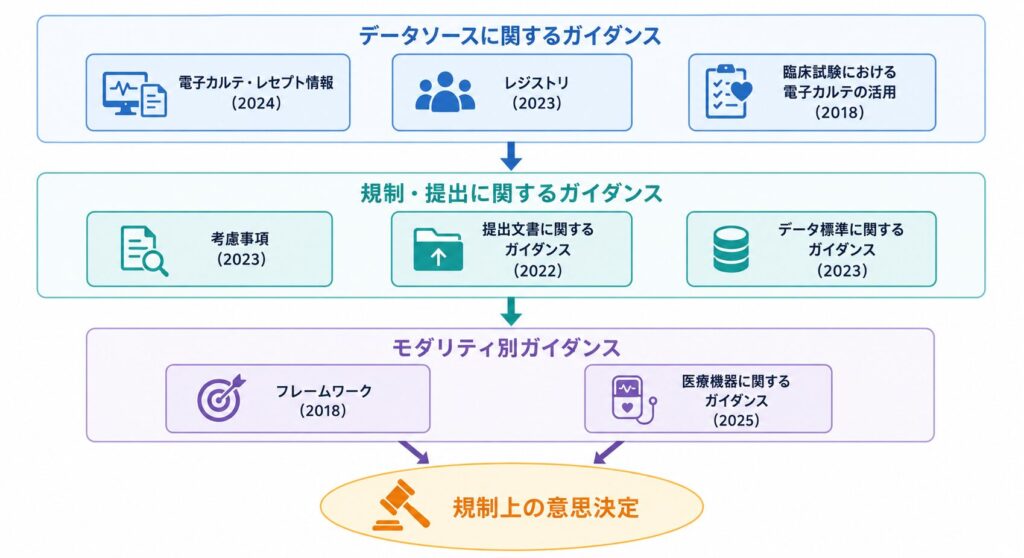
- FDAが発行する RWE関連ガイダンス8本 の発行年・ステータス・対象
- データソース別ガイダンス(EHR/Claims・Registries・EHR Clinical Investigations)の使い分け
- 規制判断・提出書類のガイダンス(Considerations・Submitting Documents・Data Standards)の実務ポイント
- 2025年12月Final化された医療機器向けRWEガイダンスの最新動向
- FDA vs PMDA・厚労省のアプローチの違いと、日米同時申請における実務戦略
はじめに
医薬品開発の現場で「リアルワールドエビデンス(RWE)」という言葉を耳にする機会は、ここ数年で劇的に増えました。米国FDAは2016年の21st Century Cures Actを起点として、医薬品・生物製剤・医療機器の規制判断にRWE/RWDを活用するための公式ガイダンス文書を、矢継ぎ早に発行しています。2026年5月時点で発行済みの主要ガイダンスは合計8本にのぼり、それぞれが「データソース評価」「規制判断」「提出書類」「データ標準」「医療機器」といった異なる切り口でRWE活用の枠組みを規定しています。
しかし、生物統計担当者・規制対応担当者として現場でぶつかるのは「どの文書を、どの場面で参照すればいいのか」という整理の難しさです。本サイトでは以前にもリアルワールドエビデンス(RWE)入門でRWEの基本概念を整理しましたが、本記事ではその次のステップとして、FDAが公表している主要ガイダンス文書群を体系的に総まとめし、各文書の位置づけ・主な要件・実務での使い分けを一枚のマップにまとめます。
製薬企業の生物統計家・規制対応担当者として、日米同時申請やグローバル開発を進める際の「ガイダンス参照リファレンス」として活用していただけるよう、PMDA・厚労省の動向との対比、実務上のチェックポイントまで踏み込んで解説します。
FDA RWEガイダンスの全体マップ ― 8本の文書を一枚で俯瞰する
ここからは、実際にFDAが公表している「ガイダンス文書群」の全体像を一気に俯瞰していきます。生物統計担当者として規制申請の場面に立つと、「どの文書を、どの目的で参照すればいいのか」が意外と整理されていないことに気付くはずです。8本の主要ガイダンスを一枚のマップに落とし込み、相互の関係性まで掴んでおきましょう。
RWEプログラムの法的根拠 ― なぜFDAはガイダンスを次々と出すのか
そもそもFDAのRWEプログラムは、2016年に成立した 21st Century Cures Act の§3022に端を発します。同法は、既承認医薬品の追加効能取得や市販後試験要件の支援に向けて、RWEを活用する枠組みを2018年12月までに策定することをFDAに義務付けました。これは連邦食品医薬品化粧品法(FD&C Act)の §505F として法典化され、PDUFA Ⅵ(2017年再認可)でもRWE活用が優先課題に組み込まれています。つまり、FDAがRWEガイダンスを矢継ぎ早に出しているのは「単なる科学的関心」ではなく、法律で明確に課せられた宿題だからなのです(参考:[FDA Real-World Evidence Program](https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence))。
フレームワーク文書(2018年12月)の位置づけ
この法的要請に応える形でFDAが2018年12月に公表したのが、「Framework for FDA’s Real-World Evidence Program」です。これは個別具体的なガイダンスというより、後続文書群の「設計図」にあたる位置づけで、RWE活用を評価する際の 3つのコア質問 を提示している点が肝です。
(a) RWDはfit-for-useか?(データソースの関連性・信頼性)
(b) 試験デザインは特定の規制上の問いに耐えうるか?(研究デザインと解析手法の妥当性)
(c) 試験運用は規制要件を満たしているか?(データ収集・モニタリング・品質保証)
後続の全ガイダンスは、この3軸のいずれかを深掘りした「分担執筆」と捉えると見通しがよくなります。
主要ガイダンス8本の一覧マップ
それでは、現時点(2026年5月)で発行済みの主要ガイダンス8本を一覧化します。発行年月とステータスはFDA公式およびFederal Registerで確認した最新情報です。
| ガイダンス文書名 | 発行年月 | ステータス | 主な対象 |
|---|---|---|---|
| Framework for FDA’s RWE Program | 2018年12月 | Framework | RWE活用の全体方針 |
| Use of EHR Data in Clinical Investigations | 2018年7月 | Final | 介入試験でのEHR活用(source data) |
| Data Standards for Drug/Biological Submissions Containing RWD | 2023年12月 | Final | RWD提出時のデータ標準(SDTM等) |
| RWD: Assessing Registries to Support Regulatory Decision-Making | 2023年12月 | Final | 疾患レジストリの適格性評価 |
| RWD: Assessing EHR and Medical Claims Data | 2024年7月 | Final | EHR・保険クレームの適格性評価 |
| Submitting Documents Using RWD/RWE to FDA for Drugs/Biologics | 2022年9月 | Final | 提出書類の識別・分類ルール |
| Considerations for the Use of RWD/RWE to Support Regulatory Decision-Making | 2023年8月 | Final | 非介入(観察)研究の規制要件 |
| Use of RWE to Support Regulatory Decision-Making for Medical Devices | 2025年12月 | Final(2017年版改訂) | 医療機器(CDRH管轄) |
ガイダンス相互の関係性 ― 3つのレイヤーで整理する
8本を眺めても、初見では「似たような名前が並んでいる」と感じるはずです。そこで、以下の3レイヤーに分けて頭の中で整理してみてください。
レイヤー① データソース系(fit-for-useの判断材料):「使おうとしているデータは本当に使えるのか?」を扱う3本がここに属します。Registries(疾患レジストリ)、EHR and Medical Claims Data(電子カルテ・保険請求データ)、そして介入試験でEHRをsource dataとして使うケースを扱う Use of EHR Data in Clinical Investigations です。データソースの種別ごとに評価軸が異なるため、扱うデータ種別に応じて参照する文書を選び分けます。
レイヤー② 規制判断・運用系(試験デザインと提出手続):「規制上の問いに答えられる試験になっているか?」「FDAに正しく提出できるか?」を扱う層です。観察研究を中心に21 CFR Part 312の適用範囲や試験運用上の責務を整理した Considerations(2023年8月)、提出書類のカバーレターでRWE使用を明示する手続を定めた Submitting Documents(2022年9月)、SDTM等のデータ標準適用上の課題を扱う Data Standards(2023年12月)の3本がここに該当します。
レイヤー③ 媒体別(医薬品 vs 医療機器):FDA内でも管轄部門が分かれており、医薬品・生物製剤はCDER/CBER、医療機器はCDRHが所管します。Use of RWE for Medical Devices(2025年12月Final、2017年版を全面改訂)はCDRH管轄で、510(k)・PMA・De Novo申請でのRWE活用を扱います。生物統計担当者が医薬品案件と機器案件を兼務している場合、この 媒体別の文書の使い分け が落とし穴になりやすいので注意が必要です。
プロトコル設計段階 → Framework + Considerations/データソース選定 → Registries or EHR&Claims/提出準備 → Submitting Documents + Data Standards/医療機器案件 → Devices(2025年12月)。この4ステップで参照文書を選べば迷いません。
データソース別ガイダンス ― EHR・医療請求データ・レジストリの評価フレームワーク
FDAのRWEガイダンス群の中でも、生物統計担当者が日々の業務で最も頻繁に参照することになるのが、データソース別のfit-for-use評価ガイダンスです。EHR(電子健康記録)、医療請求データ(claims)、患者レジストリといった異なる性質を持つRWDをどのように評価し、規制申請に耐えるエビデンスへと昇華させるか。本章では、FDAが公表している3本のデータソース別ガイダンスを横断的に整理し、それぞれの位置づけと実務上の使い分けを解説します。
Real-World Data: Assessing Electronic Health Records and Medical Claims Data(2024年Final)
2024年にFinal化された本ガイダンスは、EHRおよび医療請求データを規制申請の根拠として用いる際の評価フレームワークを示したものです。生物統計担当者にとって最も実務的なリファレンスの一つと言えるでしょう。
ガイダンスの中核は、データの relevance(関連性) と reliability(信頼性) という二軸での評価です。Relevanceとは、研究目的・対象集団・曝露定義・アウトカム定義に対して、当該データソースが必要な情報を保持しているかどうかを問うものです。たとえば抗がん剤の有効性評価において、EHRに腫瘍縮小効果(RECIST評価)が体系的に記録されていなければ、そのデータソースはfit-for-useとは言えません。
Reliabilityでは、データの 完全性(completeness)・一貫性(consistency)・正確性(accuracy)・最新性(timeliness) が問われます。請求データは「請求のため」に作成されており、診断コードの精度や記録の完全性に固有のバイアスが存在することを前提に評価する必要があります。
本ガイダンスでは、OMOP CDMやSentinel CDMといった共通データモデル(Common Data Model)の活用が推奨されています。CDMにより、複数のデータソースを統一的な構造で扱えるため、再現性・透明性が大きく向上します。また、アウトカム定義の妥当性を担保するための検証研究(validation study)の実施、データキュレーションプロセスとデータ抽出仕様書(data extraction specification)の整備が、FDAレビュー対応において必須事項となります。
Real-World Data: Assessing Registries to Support Regulatory Decision-Making(2023年12月Final)
2023年12月にFinal化されたレジストリガイダンスは、患者レジストリ・疾患レジストリを規制エビデンスの基盤として活用する際の要件を整理しています。希少疾患領域や長期安全性評価において、レジストリは依然として極めて重要なデータソースです。
評価の出発点は レジストリの目的設計 にあります。当該レジストリが「何のために」「どの集団を対象に」設計されたかを明確にし、研究目的との整合性を確認することが求められます。登録基準(inclusion criteria)、データ収集プロトコル、データ要素の選定、アウトカム定義の妥当性が、いずれも評価対象です。
加えて、患者プライバシーと倫理審査体制、長期的な ガバナンス(governance) の持続可能性も重要な評価軸となります。レジストリは数年から数十年単位で運用されるため、データの品質管理体制、資金的・組織的な継続性が担保されているかは、FDAが特に注視するポイントです。
近年は、レジストリデータを 外部対照群(External Control Arm) として活用する事例が増加しています。本ガイダンスは、Target Trial Emulationの枠組みでレジストリを活用する際の前提条件を整理する役割も果たしています。
Use of Electronic Health Record Data in Clinical Investigations(2018年7月Final)
2018年に公表された本ガイダンスは、前述の2本とは趣旨が異なる点に注意が必要です。本ガイダンスは、従来の介入型臨床試験(traditional interventional trial)において EHRデータを活用する場合の要件を定めたものであり、観察研究としてのRWE評価とは文脈が異なります。
ただし、実務的には pragmatic trial(プラグマティック試験) の基礎となる重要なガイダンスです。EHRを直接的に 電子症例報告書(eCRF) として活用する場合や、EHRから自動的に試験データを抽出する仕組みを構築する場合には、本ガイダンスの要件を満たす必要があります。
具体的には、21 CFR Part 11(電子記録・電子署名)への対応、データの完全性と原資料(source data)との整合性、被験者からのインフォームドコンセント取得プロセスなどが論点となります。
2024年のEHR/Claimsガイダンス、2023年のレジストリガイダンスは「観察研究としてのRWE」を対象とするのに対し、2018年のEHRガイダンスは「介入型試験でEHRを使う」ためのガイダンスです。社内協議や規制当局との事前相談(Type Cミーティング等)で参照を取り違えると議論が噛み合わなくなるため、目的に応じた使い分けが必須です。
3本の使い分け
3本のガイダンスの位置づけを整理すると、以下のようになります。
| ガイダンス | 対象データ | 主な評価軸 | 代表的な活用シーン |
|---|---|---|---|
| EHR/Claims(2024) | EHR・医療請求データ | Relevance/Reliability、CDM活用、検証研究 | 適応拡大、外部対照群、市販後安全性 |
| Registry(2023) | 患者・疾患レジストリ | 目的設計、登録基準、ガバナンス、倫理 | 希少疾患、長期安全性、外部対照群 |
| EHR in CT(2018) | 介入試験で使うEHR | 21 CFR Part 11、原資料整合、IC取得 | Pragmatic trial、eCRF直接連携 |
実務上は、申請目的に応じて主軸となるガイダンスを選び、補完的に他のガイダンスを参照する形が一般的です。特に、外部対照群を構築する場合は、データソース別ガイダンスとあわせて、関連する観察研究ガイダンス・外部対照群ガイダンスを併読することが推奨されます。
RWDを使った外部対照群の設計や傾向スコア分析の実装については、本サイトのリアルワールドエビデンス(RWE)入門やTarget Trial Emulation(TTE)とは、傾向スコア分析(Propensity Score Analysis)でも詳しく解説しています。
規制判断・提出書類のガイダンス ― 観察研究の規制対応からデータ標準まで
RWEを用いた申請を実務で進めるとき、生物統計担当者が最初にぶつかるのは「サイエンス」ではなく「規制手続き」と「提出フォーマット」です。いくらPS解析やTarget Trial Emulationを精緻に組んでも、INDが必要か否かの判断を誤ったり、CDISC変換が間に合わなければ、申請パッケージはそこで止まります。本章では、RWEパッケージの「ガワ」を規定する3本のガイダンス ― Considerations(規制判断の枠組み)、Submitting Documents(提出時のカバーレター)、Data Standards(データ標準)― を整理します。
3本は「規制判断(Considerations)→ 提出書類の書き方(Submitting Documents)→ 提出データの形式(Data Standards)」という申請プロセスの順番で位置づけられます。サイエンスのガイダンス群(EHR/Claims、Registries、外部対照など)が「中身」を規定するのに対し、この3本はパッケージの「枠」を規定する位置づけです。
Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drug and Biological Products(2023年8月Final)
FD&C Act 505F条項に基づき発行されたFDAのRWEプログラムの中核ガイダンスです。2021年12月のDraftを経て、2023年8月にFinal化(Federal Register 2023-08-31公示)されました。新適応取得や市販後試験要件をRWEで支援する場合の規制上の枠組みを定めています。
最大の論点は 21 CFR Part 312(IND要件)の適用可否 です。原則として観察研究(non-interventional study)はIND対象外ですが、製品の用法・用量・投与経路を意図的に変更する場合や、ランダム割付を行う場合などはINDの適用対象となり得ます。「電子カルテのデータを後ろ向きに解析するだけだからIND不要」という単純な線引きでは済まないため、プロトコル設計時点でのFDA事前相談が推奨されます。
また、観察研究であっても以下の規制上の期待が明示されました。
- 透明性の確保:プロトコルと統計解析計画の事前公表(pre-specification)、ClinicalTrials.govへの登録
- データアクセス:FDAが原データ(patient-level data)にアクセス可能であることの担保
- 試験モニタリングとデータ品質管理:プロトコル違反対応・記録保持を含むスポンサー責任
- 安全性報告:INDが必要な場合は21 CFR 312.32(IND safety reporting)に準じた手順
加えて、Final化の過程で EUA(Emergency Use Authorization)下でのRWD活用 に関する記述が追加されたのも特徴です。COVID-19パンデミック下でのRWE活用経験を反映し、Food and Drug Omnibus Reform Act of 2022(FDORA)の要請に応えた追記です。
「観察研究だからIND不要」と判断してプロトコル事前公表を怠ると、規制レビュー段階で「ポストホック解析と区別できない」と評価され、エビデンスの説得力が大幅に下がります。INDの要否にかかわらず、解析計画の事前固定とClinicalTrials.gov登録は事実上必須と考えてください。
Submitting Documents Using Real-World Data and Real-World Evidence to FDA for Drug and Biological Products(2022年9月Final)
2019年5月のDraftを経て、2022年9月にFinal化された手続きガイダンスです。狙いは明確で、FDAが「どの提出にRWD/RWEが使われたか」を内部追跡できるようにするためのカバーレター記載要件 を定めています。
具体的には、Form FDA 356h(NDA/BLA提出フォーム)に加え、カバーレターに以下を明示します。
- RWD/RWEの使用目的(safety/effectiveness のどちらを支援するか)
- 使用した研究デザイン(外部対照試験、ハイブリッド試験、レジストリ研究 等)
- 使用したRWDソース(EHR、claims、registry、product/disease registry 等)
- サブミッション分類(IND/NDA/BLA/sNDA/sBLA)ごとの記載項目
このメタデータが揃うことで、FDA側はRWEの規制活用実績を横断的に集計し、政策評価やガイダンス改訂のフィードバックループに使えるようになります。
このガイダンスは内容そのものよりも「申請パッケージの目印を付ける」手続きが主眼です。社内のレギュラトリー部門と統計部門で、どの試験を「RWE used to support a regulatory decision」と分類するかの判定基準を事前に擦り合わせておくと、提出直前の混乱を避けられます。
Data Standards for Drug and Biological Product Submissions Containing Real-World Data(2023年12月Final)
2021年10月のDraftを経て、2023年12月にFinal化されたデータ標準ガイダンスです。RWDを通常の臨床試験データと同等の電子形式で提出するための実装ルールを定めています。
中心となるのは CDISC標準 の適用です。SDTM(Study Data Tabulation Model)、ADaM(Analysis Data Model)、Define-XML を使い、EHRやclaimsといった非定型のRWDも、最終的にはSAS XPT(v5)形式に変換して提出します。eCTD(電子コモンテクニカルドキュメント)におけるRWDセクションの配置や、データ辞書・コードリスト・マッピング仕様書の整備も明示的に求められます。
注目すべきは、Final化時に 「現行のデータ標準ではRWDソースを完全には表現しきれない場合がある」 という課題認識が明文化された点です。実装上の困難については、FDAのStudy Data ResourcesやType Cミーティングを通じた事前相談が推奨されています。また、医療情報交換規格である FHIR(Fast Healthcare Interoperability Resources) との関係性にも触れられており、EHR由来データの取り扱いについては将来的にFHIRベースの規格整備が進む可能性が示唆されています。
3本ガイダンスの使い分け
| ガイダンス | 目的 | 対象 | 主な対応事項 |
|---|---|---|---|
| Considerations(2023/8 Final) | RWEによる規制判断の枠組みを定義 | 新適応取得・市販後試験要件をRWEで支援するスポンサー | INDの要否判断、プロトコル事前公表、データアクセス確保、安全性報告 |
| Submitting Documents(2022/9 Final) | RWE使用の提出をFDAが内部追跡できるようにする | IND/NDA/BLA/sNDA/sBLAでRWEを使用するすべての申請者 | カバーレターへの目的・デザイン・データソースの明示、Form FDA 356hへの追記 |
| Data Standards(2023/12 Final) | RWDを通常臨床試験データと同等の電子形式で提出 | EHR・claims等の非定型RWDを申請に使うスポンサー | SDTM/ADaM/Define-XML変換、eCTD配置、データ辞書・マッピング仕様書整備 |
実務上は、プロジェクト立ち上げ時に Considerations でIND要否と規制パスを決め、解析計画策定時に Data Standards でCDISC変換の工数を見積もり、提出直前に Submitting Documents に従ってカバーレターを整える、という時系列で運用するのが現実的です。サイエンス側のガイダンスとあわせて、申請パッケージの「枠」と「中身」を両輪で整備していきましょう。
医療機器向けRWE ― 2025年12月Final更新の最新トピック
医薬品分野のRWEガイダンスと並行して、FDAは医療機器領域でも大きな動きを見せました。2025年12月18日、Federal Registerにおいて 「Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices」のFinal Guidance が公示され、2017年版を 完全に置換(supersede) したのです。生物統計家としては、この更新が単なる文言整備ではなく、データ要件そのものの根幹を変えた点を必ず押さえておく必要があります。
2017年版からの主な変更点
最大の変更は、提出データに関する要件の緩和です。2017年版では「individual patient levelのprivate, confidential data」を求めており、結果として大規模データベース由来の macro-levelデータ(集計データ・脱識別データ) の活用が事実上困難でした。Final Guidanceではこの要求が撤廃され、de-identified data・aggregate data が許容されることが明示されています。
2025年Final化の本質は「個別患者単位データ要件の撤廃」です。これにより、claims databaseやレジストリの集計データ、合成データセットなど、これまで医療機器申請で活用しづらかったデータソースが正面から使えるようになりました。法的根拠は2022年12月のFDORA(Food and Drug Omnibus Reform Act)第3629条にあり、FDAへの明確なRWE推進ミッションが付与されています。
評価フレームの考え方も整理されました。FDAはRWDが「regulatory-grade」のRWEを生成できるかを判断する際、relevance(研究目的との整合性) と reliability(accrual・accuracy・completeness) の二軸で評価する立場を改めて明示しています。Relevanceでは対象患者集団・暴露・アウトカム定義との整合、Reliabilityではデータ取得プロセスの追跡可能性と完全性が問われます。
申請類型別のRWE活用方針
医療機器申請は薬剤と異なり、510(k)・De Novo・PMAなど複数の経路があり、各経路でのRWE活用の重みが異なります。Final Guidanceは、特に PMA(市販前承認) でのpivotal evidenceとしてのRWE利用、De Novo での実質的同等性の補強、510(k) でのpredicate deviceとの比較における補助的データとしての位置づけを整理しています。
実務上は、申請前の Pre-submission(Q-Sub)プロセス でRWE活用計画をFDAと事前協議することが強く推奨されています。研究目的・データソース・解析計画をプロトコル段階で擦り合わせることで、申請後のリジェクトリスクを大幅に下げられます。
薬剤向けRWEガイダンスとの相違点
ここで、薬剤・生物製剤向けのRWEガイダンス群と医療機器向けFinal Guidanceの違いを整理しておきます。
| 比較軸 | 医療機器向け(2025 Final) | 薬剤・生物製剤向け |
|---|---|---|
| 適用条文 | FD&C Act §513・519・520、FDORA §3629 | 21st Century Cures Act §3022、FD&C Act §505F |
| データ要件 | de-identified / aggregate data許容、macro-level活用可 | 個別患者レベルデータが基本(追跡可能性重視) |
| 評価軸 | relevance / reliability の二軸+fit-for-purpose | relevance / reliability に加え、因果推論のrigor・バイアス制御を強調 |
| 主な提出経路 | 510(k) / De Novo / PMA / HDE / IDE | IND / NDA / BLA / sNDA(適応拡大での活用が中心) |
| 事前相談 | Q-Sub(Pre-submission)で計画段階から協議 | Type B / Type C meetingで個別協議 |
医療機器向けFinal Guidanceの考え方を、そのまま薬剤・生物製剤申請に流用してはいけません。特に「aggregate data許容」は医療機器領域に固有の判断であり、薬剤領域では依然として個別患者レベルデータと追跡可能性が原則です。両ガイダンスは目的・適用条文・評価ロジックが異なるため、開発品目の規制カテゴリーを正しく見極めたうえで適用するガイダンスを選択してください。
なお、FDAは本Final Guidanceの運用開始にあたり、公示後60日程度のオペレーション期間を想定しており、すでに係属中の申請や公示直後の申請については新ガイダンスの内容を直ちに反映することを必須としていません。経過措置を踏まえつつ、次回サブミッションから本ガイダンスに沿った構成へ移行する計画を立てるのが現実的です。
日本(PMDA)の動向と実務でのポイント
FDAのRWEガイダンス群が大きく前進する一方、日本でもPMDA・厚生労働省を中心にRWE/RWDの薬事利用が着実に拡大しています。生物統計担当者は、グローバル開発を進めるにあたって日米の制度差分を正しく把握し、ICH-M14時代の国際整合も視野に入れて戦略を立てる必要があります。
PMDA・厚労省の主要文書と制度整備
日本の制度整備の流れを整理すると、以下の3つの軸で進展してきました。
第一は GPSP省令(医薬品の製造販売後の調査及び試験の実施の基準に関する省令)の2018年4月改正 です。これにより、従来は医療機関から収集した個別データに限られていた製造販売後調査に、「製造販売後データベース調査」 が新たに加わりました。レセプト・DPC・電子カルテ由来のデータベース、および疾患レジストリを再審査資料として活用する道が制度的に開かれた点が画期的です。
第二は 2021年3月の厚労省2文書 です。「承認申請等におけるレジストリの活用に関する基本的考え方について」と「レジストリデータを承認申請等に利用する場合の信頼性担保のための留意点について」が同時発出され、有効性・安全性評価に向けたレジストリ利用の枠組みと信頼性担保要件が示されました。さらに 2022年9月14日付の事務連絡(Q&A) で、レジストリ・医療情報データベースを承認申請・再審査等に利用する際の信頼性担保に関する具体的な質疑応答が示され、実務適用が進めやすくなっています。
第三は PMDA運営のMID-NET の活用拡大です。9拠点・約500万人規模の医療情報データベースで、早期安全性シグナル検出運用、NCDA(国立病院機構データ)との連携など、薬事審査・安全対策双方での利用が広がっています。
FDA vs PMDA の対比
両規制当局のRWE/RWDアプローチには、共通点と相違点があります。生物統計実務では、この差分を踏まえた dual-track戦略 が求められます。
| 比較軸 | FDA | PMDA・厚労省 |
|---|---|---|
| 規制枠組みの根拠 | 21st Century Cures Act §3022、FDORA §3629、PDUFA関連 | 薬機法、GPSP省令、厚労省通知(2021年基本的考え方ほか) |
| 主なデータソース | claims(Medicare等)、EHR、レジストリ、Sentinel | MID-NET、NDB、DPC、レセプトデータ、疾患レジストリ |
| データ標準 | SDTM/ADaM(CDISC)、OMOP CDM、Sentinel CDM | SDTM/ADaM準拠を基本、MID-NET標準フォーマット、レジストリ個別仕様 |
| 主な申請利用領域 | 適応拡大、external control、製造販売後安全性、希少疾患 | 再審査(製造販売後DB調査)、希少疾患の承認申請、外部対照群 |
| 事前相談 | Type B/C meeting、Q-Sub(機器) | PMDA対面助言(レジストリ活用相談を含む)、事前面談 |
| 信頼性担保の特徴 | relevance / reliability の二軸、protocolのpre-specification重視 | 適合性・信頼性の二軸、SOP整備とトレーサビリティを厳格運用 |
実務でのチェックポイント
最後に、生物統計担当者・規制対応担当者がRWE活用プロジェクトを進める際に必ず押さえておきたい実務ポイントを整理します。
- プロトコルの事前公表:研究目的・主要評価項目・解析計画をデータロック前にClinicalTrials.gov・jRCT・UMIN等で登録し、post-hoc批判を回避する
- fit-for-use評価:データソースのrelevance(対象患者・暴露・アウトカム定義の整合)とreliability(accrual・accuracy・completeness)を、研究目的ごとに事前評価する
- IND/治験届の要否判断:FDAでは介入を伴うRWE研究はIND範疇となる場合があるため、研究デザイン確定前に確認する。日本でもプロトコル次第で治験届該当性を要検討
- データ標準対応:FDAではCDISC(SDTM/ADaM)準拠が必須。PMDAでも国内申請でCDISC対応が広がっており、レジストリデータをマッピング可能な構造で設計する
- 事前相談の活用:PMDA対面助言・事前面談、FDA Type B/C meeting・Q-Subを早期に活用し、データソース選定段階から規制当局と合意形成を進める
RWE活用プロジェクトは「データを集めてから解析計画を考える」進め方では成功しません。プロトコル先行・fit-for-use事前評価・規制当局との早期コンセンサスの3点セットが成否を分けます。特に日米同時申請を狙う案件では、MID-NETやSentinelなど各国主要データベースの仕様差を踏まえ、解析計画書(SAP)段階で両国の要求を満たす設計に統一しておくことが、後戻りを防ぐ最大の武器になります。ICH-M14の動向も並行ウォッチし、国際整合の波に乗り遅れないよう備えましょう。
この記事をより深く理解するための参考書籍
RWE・薬剤疫学・臨床試験の規制対応をさらに学びたい方に、おすすめの書籍をご紹介します。






関連記事・次のステップ
本記事の理解をさらに深めるために、本サイトの以下の関連記事もあわせてご覧ください。
- リアルワールドエビデンス(RWE)入門 ― 規制活用と外部対照群への展開
- Target Trial Emulation(TTE)とは ― RWDで仮想ランダム化試験を再現
- 外部対照群(External Control Arm)とは ― FDA・PMDA規制動向
- 傾向スコア分析(Propensity Score Analysis)とは
- ICH E9(R1) Estimandフレームワーク徹底解説
まとめ
本記事では、FDAが発行している Real-World Evidence (RWE) 関連の主要ガイダンス8本 を体系的に整理し、それぞれの位置づけ・要件・実務での使い分けを総まとめしました。21st Century Cures Act §3022を起点とするFDA RWEプログラムは、2018年12月の Framework を皮切りに、データソース別(EHR/Claims・Registries・EHR Clinical Investigations)・規制判断系(Considerations・Submitting Documents・Data Standards)・医療機器向け(2025年12月Final)と段階的に整備が進められ、2026年時点で「医薬品・生物製剤・医療機器の全領域でRWEを規制判断に活用する」基盤がほぼ完成した状況です。
生物統計担当者として重要なのは、各ガイダンスを 「いつ」「どの場面で」参照するか を明確に理解し、プロトコル設計段階からfit-for-use評価・データ標準対応・規制当局との事前相談を計画的に組み込むことです。特に日米同時申請を視野に入れる案件では、FDAガイダンスとPMDA・厚労省通知の差分を踏まえたdual-track戦略が、成否を分ける鍵となります。
FDAはすでに「次の波」としてICH-M14(医薬品安全性評価における薬剤疫学的検討の計画・デザイン・解析)の整備にも参画しており、グローバルなRWE活用は今後さらに加速していきます。本記事で示した8本のガイダンスを起点に、最新動向を継続的にウォッチしながら、現場で活用できる知見へと深化させていただければと思います。RWEを使いこなせる生物統計家こそが、これからの医薬品開発を牽引する大きな強みになります。