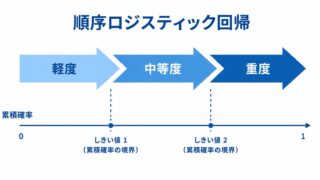
バイオ・クリープ(biocreep)とは?― ICH E10で読み解く非劣性試験の落とし穴 ―

💡 この記事でわかること
- バイオ・クリープ(biocreep)という現象の意味と、なぜそれが製薬実務において問題になるのか
- 非劣性試験を繰り返すことで、治療効果が世代を経るごとに徐々に目減りしていくメカニズム
- ICH E10ガイドラインが示す対照群選択の考え方と、バイオ・クリープとの深い関係
- 製薬企業の実務担当者がバイオ・クリープを防ぐために押さえておくべきポイント
はじめに
近年、新薬開発の現場では、プラセボ(偽薬)を対照とした臨床試験を実施することが倫理的に難しくなっています。すでに有効な治療薬が存在する疾患領域では、「効かないかもしれない薬」を患者さんに割り付けること自体が倫理上許されないケースが増えているためです。その結果、既存の標準治療薬を対照に据える「実薬対照非劣性試験(non-inferiority trial)」が、新薬開発における主流の選択肢となってきました。
非劣性試験は、「新しい薬が既存の薬に対して大きく劣ってはいない」ことを示すための試験デザインです。プラセボ対照試験のように「効くか効かないか」をゼロから検証する必要がないため、現実的で倫理的にも受け入れられやすい設計といえます。
しかし、ここに大きな落とし穴があります。非劣性試験を世代をまたいで繰り返していくと、本来の治療効果が知らないうちに少しずつ目減りしてしまい、最終的にはプラセボと変わらない薬が承認されてしまうリスクがあるのです。このリスクこそが、本記事のテーマである「バイオ・クリープ(biocreep)」と呼ばれる現象です。
本記事では、統計学を本格的に学んだことがない製薬関係者の方にも理解いただけるよう、図解と身近な例えを交えながら、バイオ・クリープのメカニズムと、それを防ぐためのICH E10ガイドラインの考え方を丁寧に解説していきます。
バイオ・クリープ(biocreep)とは何か
バイオ・クリープという言葉は、「biology(生物学・生体)」と「creep(じわじわと進む、忍び寄る)」を組み合わせた造語です。文字通り、生物学的な効果がいつの間にかじわじわと劣化していく現象を指します。
定義を整理しておきます。バイオ・クリープとは、新しい薬を「直前世代の薬」と非劣性比較していくことを繰り返すうちに、世代を経るごとにわずかな効果低下が積み重なり、最終的にはプラセボと変わらない(あるいはプラセボに劣る)薬が承認されてしまう現象を意味します。
このたとえ話としてわかりやすいのが、コピー機の世代コピーです。原本(プラセボに対してしっかり効果が確認された第1世代薬)を一度コピーした紙は、原本とほとんど見分けがつきません。しかし、そのコピーをさらにコピーし、そのまたコピーを取り続けると、いつの間にか文字がぼやけ、最終的には何が書かれているのかわからなくなってしまいます。バイオ・クリープも、これと同じように「ほんのわずかな劣化」が世代をまたいで蓄積していく現象だとイメージしていただくとわかりやすいでしょう。
製薬業界においてバイオ・クリープが問題視されるのは、次のような複数の観点があるためです。規制当局の視点では、承認した薬の臨床的価値が担保できなくなり、医療制度全体の信頼性が揺らぎます。倫理的な観点からは、本来であれば「効果があるはず」と信じて投与された薬が、実際にはほとんど効いていない可能性が出てきます。そして何より、最も大きな影響を受けるのは患者さんご自身です。効果がほとんど期待できない薬を高い薬価で服用し続けることになりかねません。
⚠️ ここに注意:「非劣性」は「同等」ではありません
非劣性試験で示されるのは、あくまで「あらかじめ決めた許容範囲(非劣性マージン)を超えて劣ってはいない」という事実だけです。つまり、新薬は対照薬とまったく同じ効果を持つわけではなく、「マージン分だけ劣っている可能性」を常に含んでいます。この“マージン分の劣化”が世代を超えて積み重なることが、まさにバイオ・クリープの本質です。
ICH E10ガイドライン(対照群選択の指針)の全体像については、以下の記事で詳しく解説していますので、あわせてご覧ください。
→ 【徹底解説】ICH E10「臨床試験における対照群の選択」
なぜバイオ・クリープが起こるのか
ここからは、バイオ・クリープがどのようにして発生するのか、そのメカニズムを段階的に見ていきます。難しい数式は使わず、シンプルな数値例で追いかけていきましょう。
まず出発点として、第1世代薬Aがプラセボとの比較で開発されたケースを考えます。ここでは、薬Aがプラセボに対して効果差Δ=10という確かな上乗せ効果を示したとしましょう。この時点では、薬Aは「プラセボより10ポイント分しっかり効く薬」として位置づけられます。
次に、第2世代薬Bの開発を考えます。すでに薬Aという有効な標準治療が存在しているため、倫理的にプラセボ対照試験は実施できません。そこで、薬Bは薬Aを対照とした非劣性試験で評価されることになります。非劣性マージンをM=3と設定したとすると、薬Bは「薬Aより3ポイント以上劣らない」ことが示されれば承認されます。ここで注意したいのは、薬Bが薬Aよりちょうど3ポイント劣っていたとしても、形式上は非劣性が成立してしまう点です。この最悪のシナリオを考えると、薬Bのプラセボに対する効果は10−3=7ポイントまで目減りする可能性があります。
そして、薬Cの開発が始まります。今度は薬Bが標準治療となっているため、薬Cは薬Bを対照とした非劣性試験で評価されます。再び非劣性マージンM=3で設計されたとすると、薬Cは薬Bよりちょうど3ポイント劣っていても承認されます。すると、薬Cのプラセボに対する効果は、最悪のケースで7−3=4ポイントまで縮んでしまうことになります。
最後にこの流れを整理すると、世代を一つ進むごとに、対プラセボの効果差は非劣性マージンの分だけじわじわと縮小していくことがわかります。下の表で、この劣化の様子を確認してみましょう。
| 世代 | 比較対照 | 非劣性マージン | 想定される対プラセボ効果差(最悪ケース) |
|---|---|---|---|
| 第1世代:薬A | プラセボ | ― | Δ = 10 |
| 第2世代:薬B | 薬A | M = 3 | Δ − M = 7 |
| 第3世代:薬C | 薬B | M = 3 | Δ − 2M = 4 |
| 第4世代:薬D | 薬C | M = 3 | Δ − 3M = 1 |
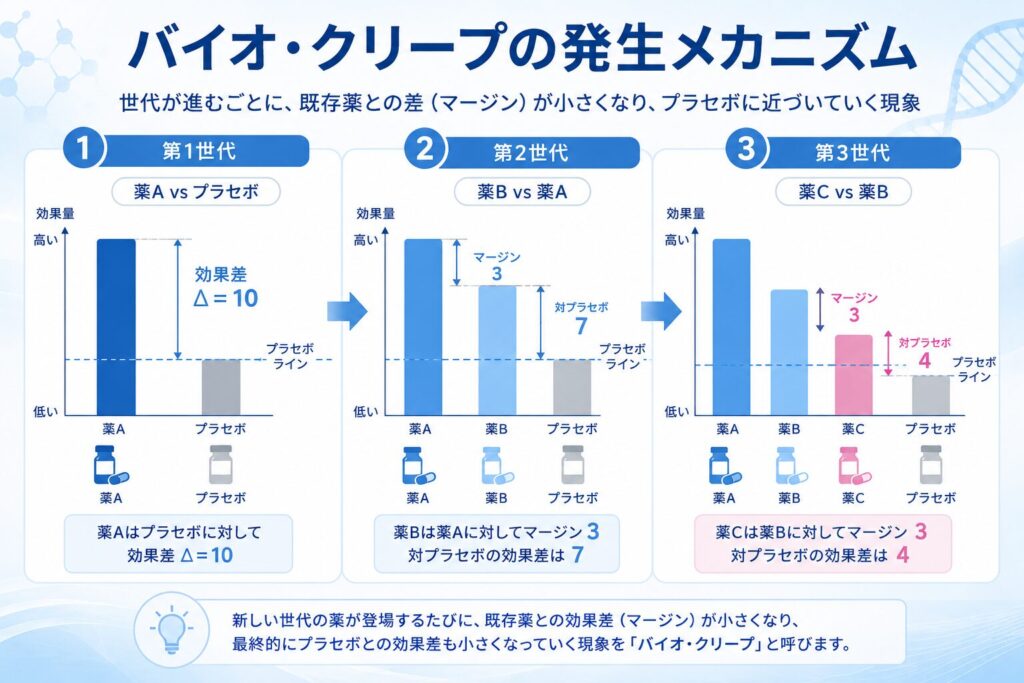
このように、わずか4世代の非劣性試験を積み重ねただけで、対プラセボの効果差は10ポイントから1ポイントへと激減してしまいます。第4世代の薬Dは、もはやプラセボとほぼ変わらない効果しか持たない可能性があるのです。
📝 補足:これは最悪ケースですが、現実に起こり得ます
もちろん、すべての世代で「ちょうどマージンギリギリ劣っている」という最悪のシナリオが連続することは、現実にはあまり多くないかもしれません。しかし、複数の非劣性試験を世代をまたいで積み重ねるなかで、各世代がそれぞれわずかに劣るパターンが少しずつ重なれば、十分に現実的なリスクとして立ち現れます。だからこそ、非劣性マージンを慎重に設定し、対照薬の選択を吟味することが極めて重要になるのです。
非劣性マージンの具体的な設定方法については、以下の記事で詳しく取り上げていますので、あわせてご参照ください。
ICH E10が示す対照群選択 ― バイオ・クリープを生む構造
非劣性試験を理解するうえで欠かせないのが、ICH E10「臨床試験における対照群の選択」(2001年発出) です。このガイドラインは、試験デザインの根幹である「対照群(コントロール群:試験薬の効果を比較するための比較対照)」をどのように設計すべきかを体系的に整理したものです。対照群の主たる目的は、試験治療の効果を疾患の自然経過や期待される効果と明確に区別することにあります。
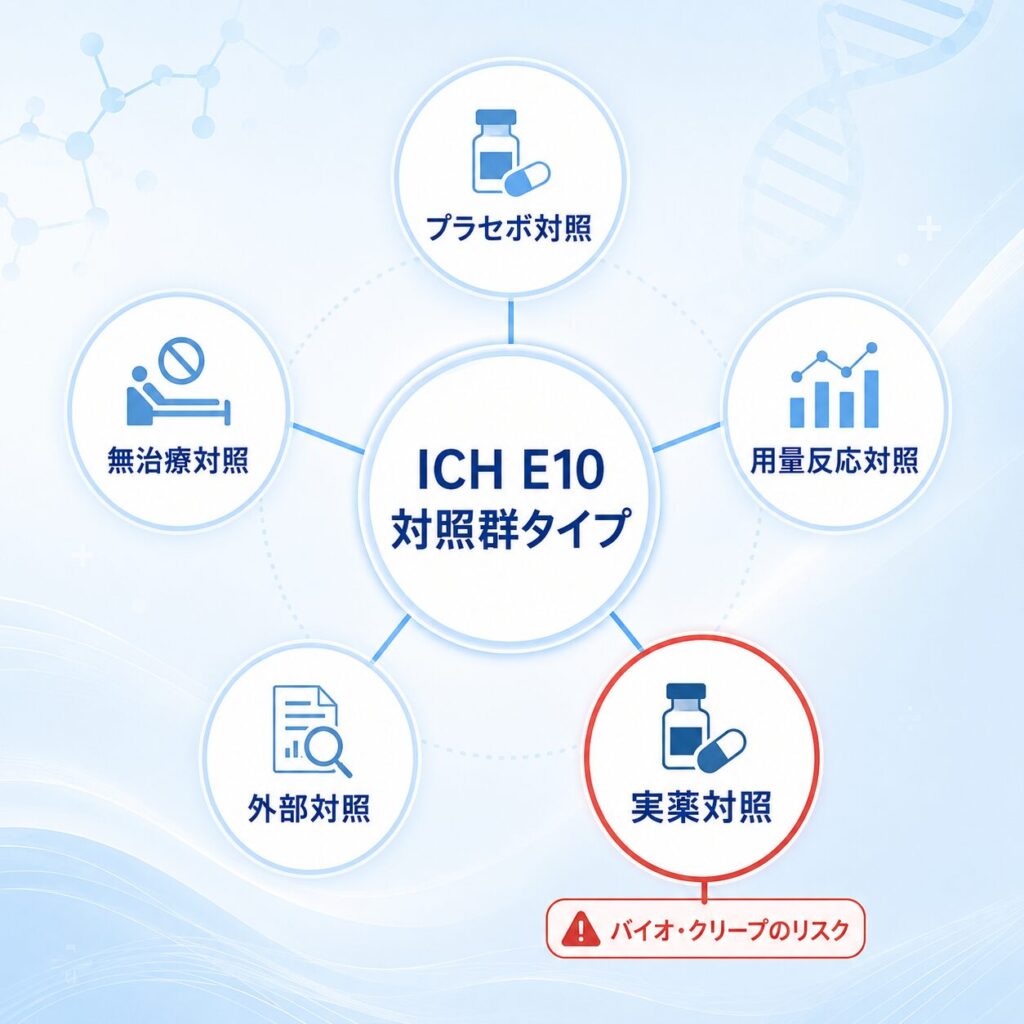
ICH E10では、対照群を以下の5つのタイプに整理しています。それぞれの特徴を整理してみましょう。
| 対照群タイプ | 概要 | 主な用途・特徴 |
|---|---|---|
| プラセボ対照 | 薬理活性のない偽薬を投与する群 | 薬剤の絶対的効果を最も明確に検出可能 |
| 無治療対照 | 何も投与しない群(盲検化はできない) | 手術や物理的介入の評価などに用いる |
| 用量反応対照 | 同じ薬の複数用量を比較する | 用量と効果の関係を検証できる |
| 実薬対照 | 既存の標準治療薬を対照とする | 倫理的にプラセボが使えない領域で頻用/非劣性試験で多用 |
| 外部対照(ヒストリカル含む) | 過去の試験データや別集団を対照とする | 希少疾患などで活用されるがバイアスが大きい |
ここで注目していただきたいのが、実薬対照(Active Control)を用いた非劣性試験には特有のリスクがあるという点です。プラセボ対照試験であれば「薬剤に効果があるか/ないか」を直接判定できますが、実薬対照の非劣性試験では、試験薬と実薬を比較するだけでプラセボとの直接比較は行いません。
ここで重要になるのが、「constancy assumption(恒常性仮定:過去の試験で観察された実薬の効果が、現在実施する試験でも同程度に保たれているという仮定)」 という概念です。もしこの仮定が崩れていれば、過去のデータを根拠に設定された非劣性マージンそのものが意味を持たなくなってしまいます。
そしてもうひとつの鍵が、「assay sensitivity(検定感度:実際に効果のある薬と効かない薬を区別する能力)」 です。試験そのものに薬剤の効果差を検出する力がなければ、「非劣性が示せた」という結果も、本当に試験薬が有効だからなのか、それとも単に差を検出できなかっただけなのか、区別がつかなくなってしまいます。
⚠️ 注意
実薬対照の非劣性試験ではプラセボとの直接比較が行われないため、constancy assumptionとassay sensitivityという2つの前提が崩れた瞬間に、結果の解釈が一気に揺らぎます。世代を重ねるごとにこれらの前提が侵食されていく状況こそが、バイオ・クリープの温床となるのです。
ICH E9との関連と「Assay Sensitivity」
非劣性試験の取り扱いは、ICH E10だけでなくICH E9「臨床試験のための統計的原則」 でも重要なテーマとして言及されています。とくにICH E9 (R1)では、推定対象(Estimand)の枠組みのなかで、非劣性試験における比較の解釈について丁寧に整理されています。
ここで改めて強調しておきたいのが、Assay Sensitivityが担保されない非劣性試験の危うさです。
例えば、ある新薬Xと標準薬Yを比較した非劣性試験で「Xは非劣性が示された」という結果が得られたとします。しかし、もしその試験のAssay Sensitivityが不十分だったとしたら、その結果は以下のどちらの状況でも同じように見えてしまいます。
- 状況1:新薬Xが標準薬Yと同等に効いており、両方ともしっかり効果を発揮している
- 状況2:新薬Xも標準薬Yもどちらも効いておらず、結果として差が出ていないだけ
統計的には「差がない」という同じ結論に見えるのですが、患者さんへの影響は天と地ほど違います。これが非劣性試験の最も怖いところであり、Assay Sensitivityを担保する設計が極めて重要になります。
この問題への対応として用いられるのが、Put-it-together(PTI)アプローチ や 仮想プラセボ(Putative Placebo)との比較 という考え方です。これは、過去のプラセボ対照試験のデータを利用して「もし今回の試験にプラセボ群があったとしたら、試験薬はプラセボに対してどの程度の効果を持っていたと推定できるか」を間接的に評価する手法です。
具体的な方法としてよく知られているのが、95-95ルール(M2法とも呼ばれる) です。これは、実薬の効果の信頼区間の下限を用い、さらに新薬の対実薬の信頼区間の下限を組み合わせて、実薬が持っていた効果のうち最低でも50%を新薬が保持していることを要求する考え方です。
📝 補足
「非劣性試験は優越性試験より楽だ」という誤解が時折見られますが、実態はむしろ逆です。プラセボ対照試験では「差があるかないか」を検証するだけでよかったところ、非劣性試験では「マージン設定の根拠」「constancy assumptionの確認」「assay sensitivityの担保」など、より厳密な事前設計と論証が求められます。「手抜き」どころか、より高度な統計的・臨床的思考が必要な試験デザインなのです。
数値で見るバイオ・クリープのシミュレーション例
ここまで概念を整理してきましたが、実際にバイオ・クリープがどのように進行するのかを、架空の降圧薬シリーズの仮想例で見ていきましょう。なお、以下の数値はあくまで概念を理解するための作例であり、実在する試験の数値ではない点をあらかじめお断りしておきます。
ある降圧薬の系統で、第1世代から第4世代まで非劣性試験を積み重ねてきたとします。各世代の試験設定は次のとおりです。
| 世代 | 直接の対照 | 非劣性マージン | 実際の効果(収縮期血圧低下) | 推定対プラセボ効果 |
|---|---|---|---|---|
| 第1世代 A | プラセボ | ―(優越性試験) | 12 mmHg(95%CI 9-15) | 約 12 mmHg(堅実) |
| 第2世代 B | A | 3 mmHg | 9 mmHg | 約 9 mmHg |
| 第3世代 C | B | 3 mmHg | 6 mmHg | 約 6 mmHg |
| 第4世代 D | C | 3 mmHg | 3 mmHg | 約 3 mmHg(プラセボと有意差なしの可能性) |
この表を眺めていただくと、各試験はそれぞれの世代ごとには「非劣性が示された」と統計的に主張できることがわかります。Bは「Aに対して3 mmHg以内の差」、Cは「Bに対して3 mmHg以内の差」、Dは「Cに対して3 mmHg以内の差」を、それぞれ満たしているからです。
ところが、プラセボに対する効果の推定値をたどっていくと、世代を重ねるごとに効果が削られていく様子が見えてきます。第1世代Aの12 mmHgから始まり、B(9 mmHg)、C(6 mmHg)と来て、第4世代Dではついに3 mmHgまで縮小しています。この水準になると、もしプラセボ対照試験を行ったとしてもDの効果はプラセボと有意差を示せない可能性が高くなります。
つまり、各試験は個別には「合格」しているにもかかわらず、シリーズ全体として見ると、いつの間にかプラセボと変わらない薬まで市場に出てきてしまうという構造になっているわけです。これがバイオ・クリープの本質です。
📝 補足
注目すべきは、非劣性マージンの設定が甘いほどバイオ・クリープが加速するという点です。上の例ではマージン3 mmHgで進行していますが、もしマージンが5 mmHgであれば、わずか2世代でプラセボと同等のレベルまで効果が削られてしまう可能性もあります。だからこそICH E10では、マージン設定の根拠を臨床的・統計的に明確に説明することが厳しく問われるのです。マージンは「都合のよい数字」ではなく、過去のプラセボ対照試験のデータと臨床的に意味のある最小効果差から論理的に導出されるべきものなのです。
バイオ・クリープを防ぐ実務的アプローチ
バイオ・クリープは「気づかぬうちに効果が目減りする」という構造的な問題です。完全に避けることは難しいものの、試験デザインと統計的工夫によってリスクを大幅に下げることができます。ここでは、製薬企業の開発現場で実際に検討されている代表的なアプローチを整理します。
非劣性マージンを保守的に設定する
非劣性マージン(new drugがactive controlに対してどの程度劣ってもよいかを示す閾値)を緩く設定してしまうと、バイオ・クリープを助長してしまいます。ICH E9(臨床試験のための統計的原則)でも繰り返し強調されているように、マージンは「臨床的に意味のある差」と「過去のプラセボ対照試験で確認された効果差」の両面から導く必要があります。
実務上よく用いられる目安としては、過去のプラセボ対照試験で示された薬剤Aとプラセボの効果差の95%信頼区間の下限値の50%以内にマージンを置く、という保守的な設定です。これは「効果の半分を失っても許容する」のではなく、「効果の半分以上は確実に残っていることを示す」というロジックに立っています。
プラセボを残せるならプラセボ群を残す
倫理的に許容される疾患領域であれば、プラセボ+実薬+新薬の3群試験を選ぶのが理想です。3群デザインであれば、実薬とプラセボの差(Assay Sensitivity、試験の感度)を同じ試験内で直接検証でき、バイオ・クリープの最大の原因である「実薬の効果が本当に出ているのか確認できない」という弱点を解消できます。
もちろん、致死的疾患や標準治療が確立している領域ではプラセボ群の設定が倫理的に困難な場合もあります。その際は、レスキュー治療の併用や短期間の観察に限定するなど、リスクを最小化する工夫が併せて検討されます。
Putative Placebo比較と95-95ルール
プラセボ群を置けない場合、過去のプラセボ対照試験のデータを使って「仮にプラセボ群があったらどうなっていたか」を推定する手法がPutative Placebo(仮想プラセボ)比較です。これは、過去試験から得られた「実薬 vs プラセボ」の効果差を、現在の試験の「新薬 vs 実薬」結果と組み合わせ、「新薬 vs 仮想プラセボ」の効果を間接的に推定するアプローチです。
また、FDAなどで参照される95-95ルールは、「過去のプラセボ対照試験で示された実薬の効果差の95%CI下限の、さらに95%CI下限を維持する」という二重に保守的な基準で、バイオ・クリープを抑える代表的な手法として知られています。承認申請時には、これらを補助的に提示することで規制当局の納得を得やすくなります。
メタアナリシスで過去試験の効果差を再確認
過去のプラセボ対照試験が複数存在する場合、メタアナリシスによって実薬とプラセボの効果差の点推定値と信頼区間を統合的に再確認しておくことが推奨されます。単一試験の結果だけを根拠にマージンを設定すると、たまたま大きな効果が出た試験に引きずられてしまうリスクがあるためです。
倫理的に許容されるなら優越性試験を選ぶ
そして根本的な対策として、可能であれば優越性試験(superiority trial)を選ぶという選択肢も忘れてはなりません。非劣性試験は「同等以上であることを示せれば良い」という設計上、どうしても基準が緩みがちです。新薬の有効性に十分な見込みがあるなら、優越性試験を選ぶことでバイオ・クリープの議論そのものを回避できます。
⚠️ 注意
「non-inferiority margin」を試験ごとに勝手に緩めてしまうと、規制当局から必ず指摘されます。マージンの根拠と設定値は、プロトコル確定前にPMDA・FDA・EMAとの事前合意(Scientific Advice、Pre-IND Meetingなど)を経ることが極めて重要です。
製薬企業の実務でのポイント
🔑 製薬実務でバイオ・クリープを抑えるための4つのチェックポイント
① 試験計画段階
非劣性マージンの根拠を、必ず歴史的データ(historical data)から定量的に示します。「臨床的に意味のある差」だけでなく、過去のプラセボ対照試験のメタアナリシス結果も併せて提示することが望まれます。
② プロトコル段階
非劣性試験では、ITT解析(intention-to-treat、割付通り解析)とper-protocol解析の両方を主要・副次解析として規定し、感度分析(sensitivity analysis)を必ず含めます。両解析で結論が一致することが、非劣性の頑健性を支える重要な要素になります。
③ 解析段階
Assay Sensitivityが確保できなかった場合の対応プランを、解析開始前に事前規定しておきます。「結果を見てから方針を変える」ことは、規制当局から最も厳しく見られるポイントです。
④ 規制対応
PMDA・FDA・EMAいずれも、非劣性試験に対しては優越性試験以上に厳しい目を持っています。マージン設定・対照薬選択・解析計画について、申請前相談(Scientific Advice / Pre-NDA Meeting)を活用し、論点を事前に潰しておくことが申請成功の鍵となります。
📚 この記事をより深く理解するための参考書籍
統計・生物統計をさらに深く学びたい方に、おすすめの書籍をご紹介します。






関連記事
バイオ・クリープの背景にあるICH E10や非劣性マージンの設定方法、効果量の考え方については、以下の関連記事もあわせてご覧ください。本記事の理解がさらに深まるはずです。
- 【徹底解説】ICH E10「臨床試験における対照群の選択」:対照群選択の原則を、Assay Sensitivityの観点から詳しく解説しています。
- 非劣性試験と非劣性マージンの設定:マージン設定の具体的な手順と、historical dataの取り扱いについてまとめています。
- 効果量(Effect Size)を理解すると統計が一気に実務的になる:マージンや効果差を考えるうえで土台となる「効果量」の考え方を解説しています。
まとめ
バイオ・クリープ(biocreep)は、非劣性試験を繰り返すうちに薬剤の真の効果がじわじわと目減りしていく、非劣性試験の構造的な落とし穴です。試験ごとに少しずつ劣る薬剤が「非劣性」と判定されて承認されていくことで、長期的には「効くかどうかも怪しい薬剤」が標準治療の座を占めてしまうリスクをはらんでいます。
この問題に対処するには、ICH E10とICH E9に基づく厳密なマージン設定と、Assay Sensitivity(試験が実薬とプラセボを区別できる感度)の確保が極めて重要になります。とくに製薬実務では、「マージンの根拠を歴史的データから明確に示すこと」「ITT・per-protocol両解析を含めた感度分析を事前規定すること」「PMDA・FDA・EMAとの事前合意を丁寧に積み上げること」――この3点が非劣性試験を成功させる鍵となります。
非劣性試験は決して「優越性試験より楽な試験デザイン」ではなく、むしろ統計的・規制的にはより慎重な設計が求められる試験です。バイオ・クリープという視点を持っておくことは、生物統計家として臨床開発に関わる際の大きな強みになります。試験計画や解析計画を検討する際の判断軸の一つとして、ぜひ役立てていただければと思います。